本文来自“单细胞组学”公众号 原文链接:https://mp.weixin.qq.com/s/ugq_JE9XOIkVDztADkp9-w
单细胞转录组数据主要存在细胞特异性和基因特异性的两类系统偏差。比如,由于测序的每个细胞样品的总量不一样,测序深度不一样,也就是文库大小不一样,这个因素是肯定需要考虑的。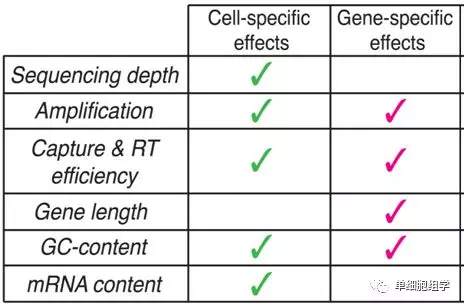
Vallejos C A etal. Nature methods 2017
标准化的目的:校正技术误差和一些不相关生物背景噪音。
1.RPM/CPM:只标准化测序深度
定义:Reads/Counts of exon model per Million mapped reads (每百万映射读取的reads)。
公式: 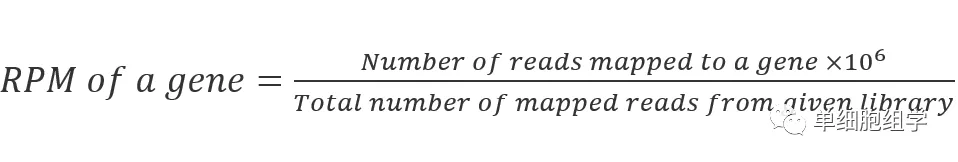
2.RPKM/FPKM:先标准化测序深度,再标准化基因长度
定义:Reads/Fragments Per Kilobase of exon model per Millionmapped reads (每千个碱基的转录每百万映射读取的reads/fragments).
公式:
3.TPM:先标准化基因长度,再标准化测序深度
定义:Transcripts Per Kilobase of exon model per Million mappedreads (每千个碱基的转录每百万映射读取的Transcripts)
公式:
4.Trimmed Meanof M-values (TMM)
定义:计算每个泳道的TMM因子,其中一个泳道被视为参考样品而其他泳道被视为测试样品。对于每个测试样品,在排除表达最多的基因和具有最大对数比的基因后,将TMM计算为该测试与参考样品之间的对数比的加权平均值。该方法是edgeR包中的标准化方法。
公式: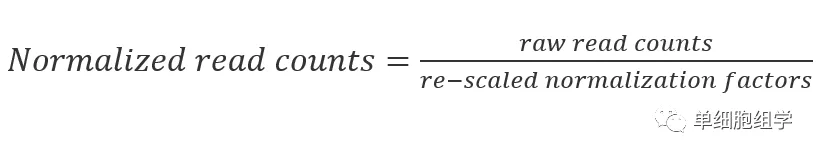
以上方法均是适用于bulk RNA-seq数据,没有考虑scRNA-seq数据的稀疏性,下面介绍一种特意为scRNA-seq数据开发的方法。
5.去卷积法,主要是为校正细胞特异性的系统误差,详见scran包的computeSumFactors函数。
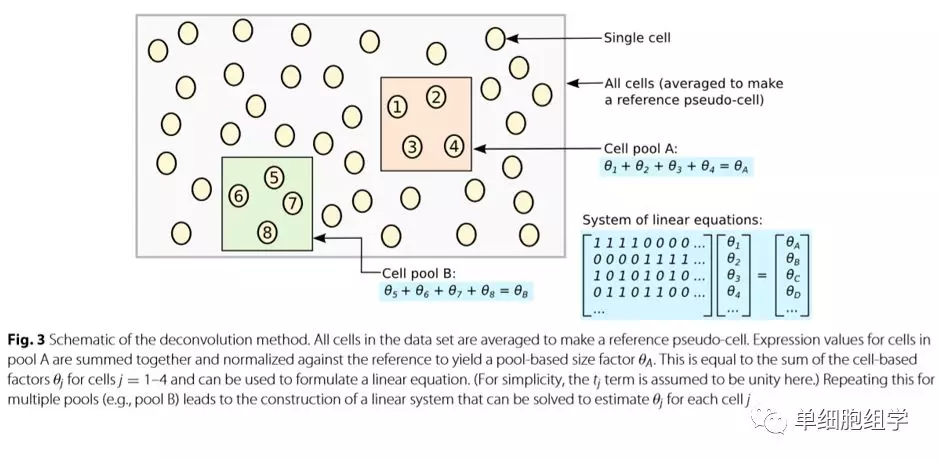 Lun A T L et al.Genome biology 2016
Lun A T L et al.Genome biology 2016
关键步骤:
1)定义一个细胞集
2)对细胞集中所有细胞的表达值求和
3)用参考假细胞对细胞集的求和表达值进行标准化
4)对多个不同的细胞集重复此操作以构建一个线性系统
5)对基于细胞集的size factors进行去卷积得到基于细胞的size factors
主要公式(具体参数解释,请参见 Lun A T L et al. Genome biology 2016):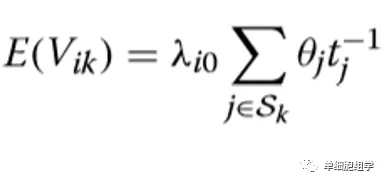
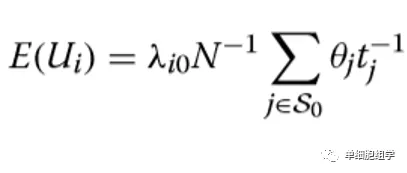
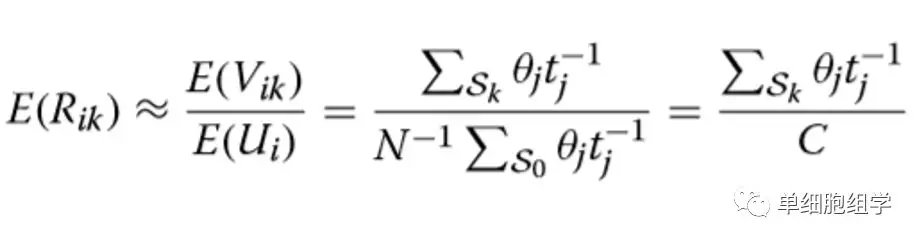
就以上介绍的几种方法而言,本文只是想通过一种浅显易懂的方式叙述它们的概念和原理,其实,在一些实际使用的生信软件中并不一定如本文所说的这么简单。标准化的方式根据实际情况调整。例如单细胞的表达量分析中,有些基因只有个位数的reads,有些成千上万,数据分布过于离散,这时可以直接应用log2(TPM+1)进行标准化。此外,目前对基因长度的定义也存在争议,有些软件是挑选基因的最长转录本,有些选取多个转录本长度的平均值等,这样对基因长度的标准化可能会引入一些人为误差。所以,对于这些标准化方法的选取,还得结合数据本身,见仁见智,毕竟能够解决科学问题的方法才是好方法。
当然,随着单细胞转录组学的迅速发展,适用于单细胞转录组数据的标准化方法层出不穷,但这些标准化方法仍需时间的检验,才能真正地被越来越多的研究人员所接受并普遍使用。
参考资料:
1. Vallejos C A, Risso D,Scialdone A, et al. Normalizing single-cell RNA sequencing data: challenges andopportunities[J]. Nature methods, 2017, 14(6): 565.
2. Lun A T L, Bach K, Marioni J C.Pooling across cells to normalize single-cell RNA sequencing data with manyzero counts[J]. Genome biology, 2016, 17(1): 75.
3. Robinson M D, Oshlack A. Ascaling normalization method for differential expression analysis of RNA-seqdata[J]. Genome biology, 2010, 11(3): R25.
4. Li B, Dewey C N. RSEM: accuratetranscript quantification from RNA-Seq data with or without a referencegenome[J]. BMC bioinformatics, 2011, 12(1): 323.
5. Trapnell C, Williams B A,Pertea G, et al. Transcript assembly and quantification by RNA-Seq revealsunannotated transcripts and isoform switching during cell differentiation[J].Nature biotechnology, 2010, 28(5): 511.
6. Butler A, Hoffman P, Smibert P,et al. Integrating single-cell transcriptomic data across different conditions,technologies, and species[J]. Nature biotechnology, 2018, 36(5): 411.

若有收获,就点个赞吧
0 人点赞
