本文来自“单细胞组学”公众号 原文链接:https://mp.weixin.qq.com/s/8lCDjXXPuNP1V7Sol-YbKA
背景
本期关注空间高变基因(spatially variable gene,SVG)统计方法,寻找具有组织空间分布特征的基因。
内容
现有的方法:
- Seurat的markvario;
- Moran’s I;
- Geary’s C;
- Trendsceek;
- SpatialDE;
- SPARK;
- SPARKX
- Giotto的binSpect.
最后挑选markvario,Moran’s I,SpatialDE,SPARK和SPARKX进行测试。
总结
- 所有方法鉴定结果相似,SVG有一半相同。
- 统计P值受spot数影响,样本spot数越多,根据P值挑选的SVG越多。
- 需要更加稳健的方法对P值进行校正。
- Seurat的方法耗时较少,SpatialDE耗时最多。
上期我们总结了单细胞转录组的高变基因(High variable gene,HVG)统计方法,本期聚焦空间转录组数据的高变基因选择方法。根据若干个空间转录组流程框架提供的信息,尽管空间转录组不同于单细胞转录组(如:1. 单细胞—>Spot;2. Spot空间位置信息),大多数分析框架的高变基因选择方法仍然与单细胞转录组相同。如基于基因表达均值和方差的回归方法等。这是不考虑空间位置信息的统计方法,主要用于后续spot的降维聚类。本期关注空间高变基因(SVG)统计方法,寻找具有组织空间分布特征的基因。
空间分析的一般流程
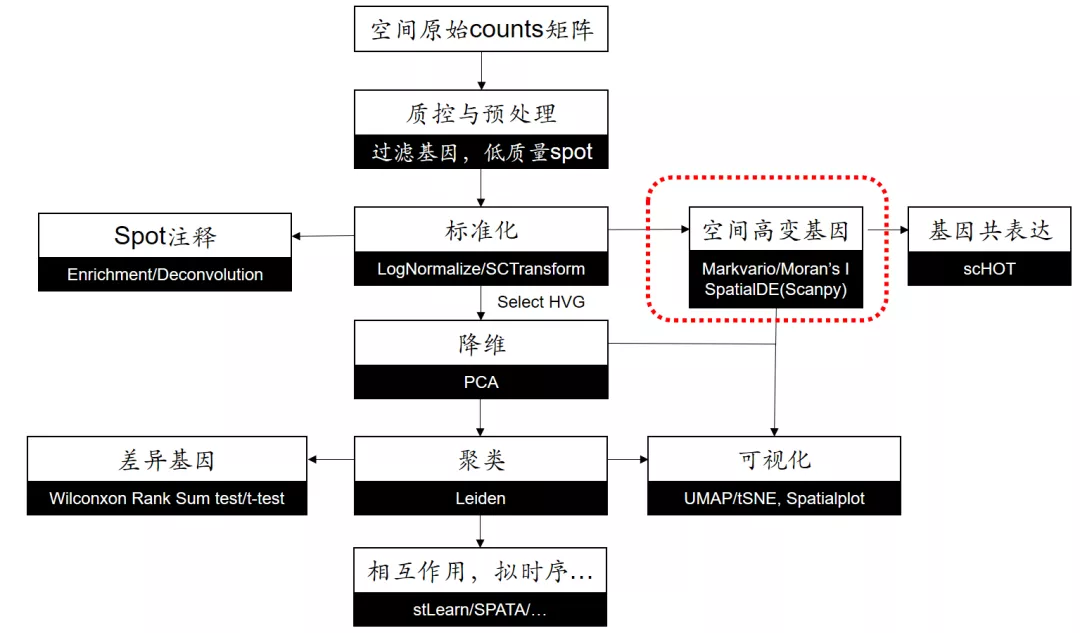
Seurat/Scanpy空转分析流程[1,2]
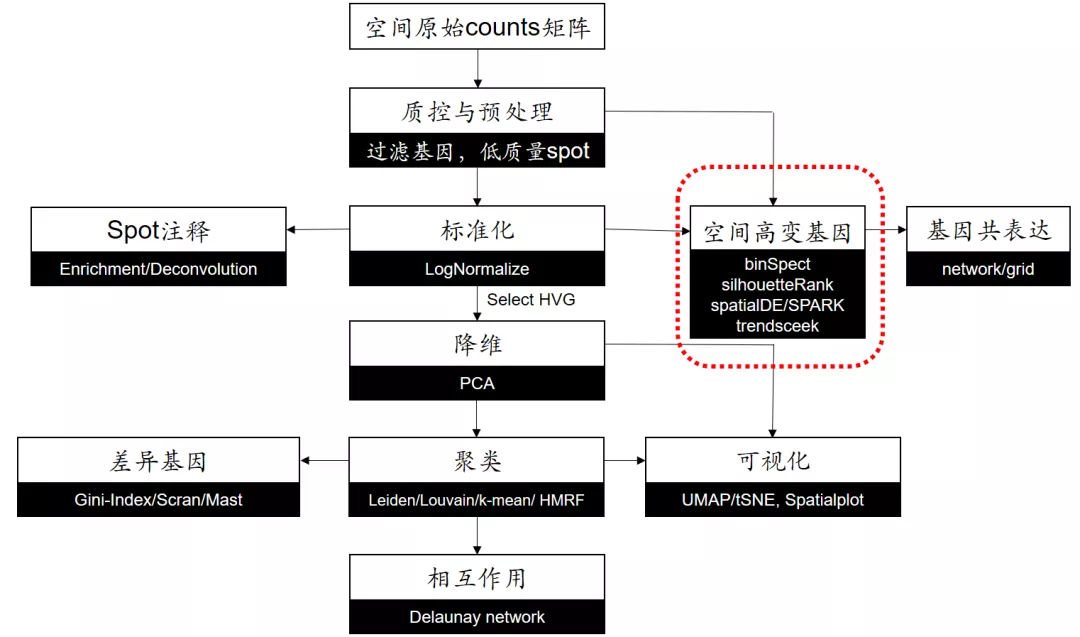
Giotto空转分析流程[3]
空间高变基因方法
自从2016年SPATIAL TRANSCRIPTOMICS的空间转录组技术问世以来,相关分析工具也如雨后春笋般涌现[4]。大多数常见的高变基因算法基于Mark variogram, 如trendsceek[5]。Mark variogram是地质学和生态学常用的空间统计算法。例如,不同树木的分布情况可以用Mark variogram衡量,选择一定范围尺度r统计,从数值大小可以看出树木是否特异聚集[6]。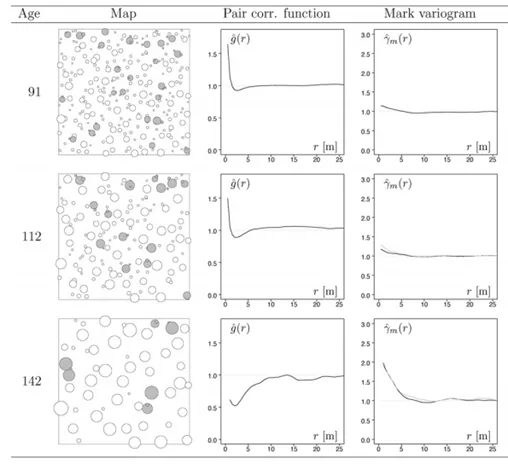
图来自文献:DOI: 10.1016/j.ecolmodel.2012.12.009
Mark variogram中的参数r可以理解为尺子的长度,如下图的红线,把尺子放在待测平面上,收集尺子两端的数值差异,计算期望值最后算出Mark variogram统计值。改变尺子长度r,可以得到Mark variogram关于r的曲线。想进一步学习可以看Michael Pyrcz的油管频道GeostatsGuy Lectures。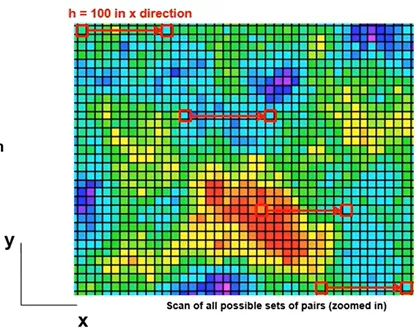
来自Michael Pyrcz[7]
Trendsceek
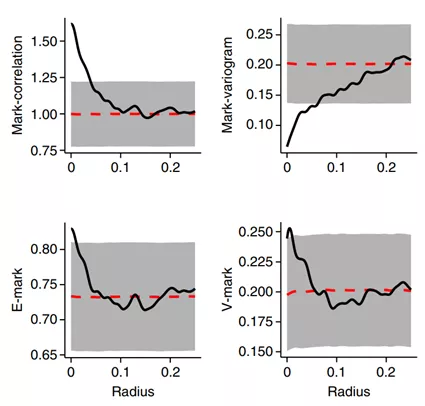
Trendsceek是2018年卡罗琳斯卡医学院Rickard Sandberg团队开发的空间高变基因方法,RickardSandberg团队也是SmartSeqV3的作者。Trendsceek综合了四种统计方法,下图是文章对Hot spot的差异基因分布类型进行量化。灰色带是随机打乱spot位置统计1000次的数值范围。如果在灰色的范围之外,表示这个基因的表达有显著的空间差异(p<0.05)。可以看出Mark-variogram和Mark-correlation用比较短的距离尺度r(radius)可以显著地表示Hot spot的空间特征。Trendsceek在github上的代码已经三年没更新了,示例代码有bug,故不测试。
markvario
Seurat的FindSpatiallyVariableFeatures函数提供两种方法选择:markvario和Moran‘s I。其中markvario方法引用R包spatstat的函数,原理和trendsceek的Mark-variogram类似。
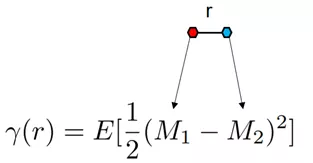
M1和M2分别是spot的表达量;
r值表示两个spotM1和M2之间的距离;
γ表示成对spot之间表达量差异平方的期望,也是我们需要获取的mark-variogram值。
可以取不同尺度的r值计算γ值: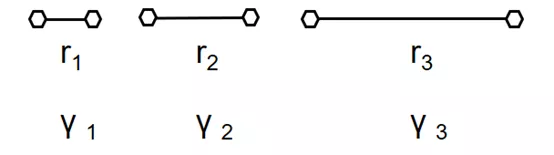
用γ的大小表示基因空间表达差异。Seurat根据r=5时的γ值,从小到大对基因进行排序,越靠前,空间差异越显著。如下图所示,基因CXCL14有显著的空间特征,当r=5时,γ值小于0.1;基因ACTB没有显著的空间特征,当r=5时,γ值接近0.65。
其他
Geary’s C也和markgram原理类似,取值范围0到2。Moran’s I原理和Pearson相关系数类似,取值范围-1到1。SpatialDE和SPARK都基于高斯过程回归(Gaussian process regression),同时考虑了spot基因表达量和spot的空间位置信息。SPARK与SpatialDE不同的是,直接在原始数据上建模,考虑了原始数据的均值和方差关系。另外SPARK对P值进行校正,减少I型错误,获得更多的空间特征基因。Giotto的方法binSpect虽然具有与SpatialDE类似的高变基因检测结果,且运行时间较短,但Giotto文章中提到还需对p值校正方法进一步优化。
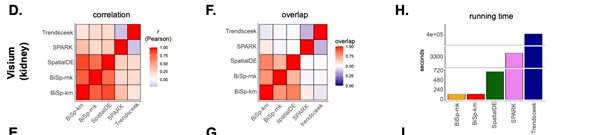
Giotto文章FigureS8. binSpect与其他方法的比较
SVG统计结果比较
接下来挑选4种方法进行测试:Seurat的markvario/Moran‘s I, SpatialDE,SPARK和SPARKX。
1. SVG比较
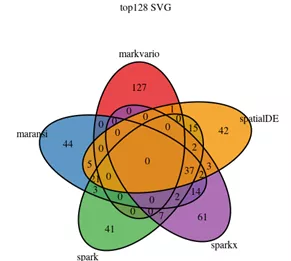
选取4种方法获得的前5000个SVG做韦恩图,可以看出所有方法共享约一半的SVG;Seurat的markvario,SpatialDE和SPARK共享3633个SVG;值得注意的是SPARKX鉴定到的1198个SVG,在其他方法中没有鉴定到。用另一个只含251个Spots的数据,得到的SVG更少。SPARK、SPARKX和SpatialDE共享37/128的SVG;SPARK、SpatialDE和Moran’s I共享21/128的SVG。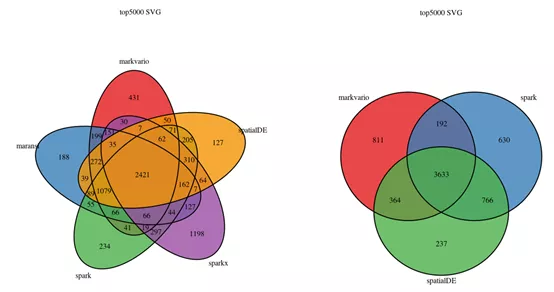
2. P值问题
除了Seurat的markvario方法直接用γ值排序之外,其他方法的统计结果都包含P值。然而大部分基因的adjust P值都很小。对所有11789基因进行比较,四种方法根据adjust P<0.05选取的SVG数目都超过10000。P值的范围似乎与数据的Spot数相关,10X visium的乳腺癌数据有3987个spot,根据P值获得的显著性SVG较多,而SpatialDE和SPARK文章用另一个数据只有251个spot,根据P值获得的显著性SVG较少。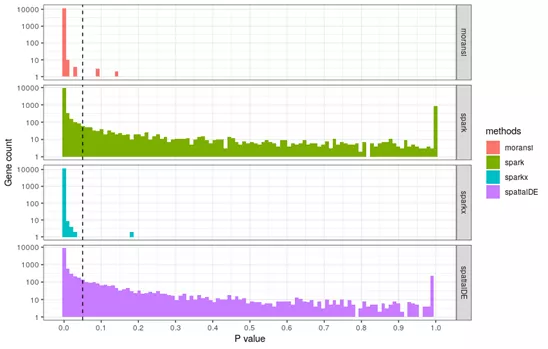
用4种方法统计乳腺癌数据SVG的P值分布
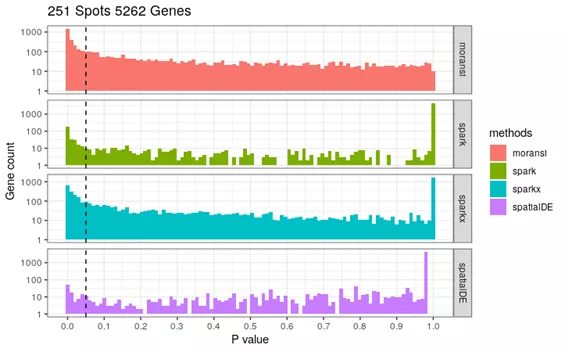
用4种方法统计较小的乳腺癌数据SVG的P值分布
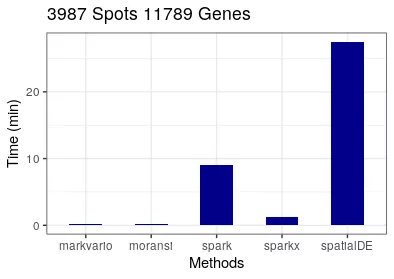
3. 运行速度
用16个线程运行两个数据的SVG分析,结果SpatialDE耗时最多,Seurat的markvario和Moran’sI耗时最少。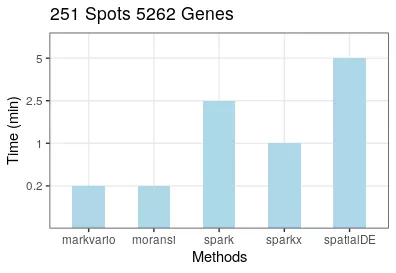
下期预告:空转算法(二):发育轨迹推断
参考链接:
[1] Butler A,Hoffman P, Smibert P, et al. Integrating single-cell transcriptomic data acrossdifferent conditions, technologies, and species[J]. Nature biotechnology, 2018,36(5): 411-420.
[2] Wolf F A, Angerer P, Theis F J. SCANPY: large-scalesingle-cell gene expression data analysis[J]. Genome biology, 2018, 19(1): 1-5.
[3] Dries R, Zhu Q, Dong R, et al. Giotto: a toolbox for integrativeanalysis and visualization of spatial expression data[J]. Genome biology, 2021,22(1): 1-31.
[4] Ståhl P L, Salmén F, Vickovic S, et al. Visualization andanalysis of gene expression in tissue sections by spatial transcriptomics[J].Science, 2016, 353(6294): 78-82.
[5] Edsgärd D,Johnsson P, Sandberg R. Identification of spatial expression trends insingle-cell gene expression data[J]. Nature methods, 2018, 15(5): 339-342.
[6] Pommerening A,Särkkä A. What mark variograms tell about spatial plant interactions[J].Ecological Modelling, 2013, 251: 64-72.
[7] https://www.youtube.com/watch?v=jVRLGOsnYuw

若有收获,就点个赞吧
0 人点赞
