本文来自“单细胞组学”公众号 原文链接:https://mp.weixin.qq.com/s/6xRy9mEr1eVgFER-AmiF9A
上周有读者反映公众号内容过于专业难懂,因此以后的内容在保持质量与专业性的同时,尽量做到通俗易懂。本期介绍常用的单细胞聚类算法之一:社区划分与模块度。
首先解释什么是单细胞组学,为什么要做单细胞。
在受到惊吓后,我们常说,“吓死了我好多脑细胞”。这个“脑细胞”其实不止一种细胞。我们的大脑可以分成多个区域,每个区域有不同比例的细胞类型,如兴奋性神经细胞,抑制性神经细胞,星状胶质细胞等。剖析脑单细胞图谱有助于脑部生物功能研究和病理探索。
单细胞组学大致可以分为基因组学、转录组学、表观和蛋白组学。目前单细胞蛋白组学仍然无法像核酸一样做到高通量高精度。最近在arXiv上有一篇做单细胞蛋白组学的文章,经过实验优化后声称可以做到一个细胞检测约1000个蛋白的较高精度,但基于TMT标签的方法只能8个细胞一组检测,无法达到高通量[1]。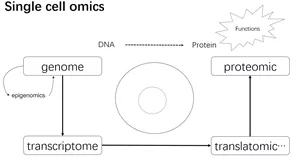
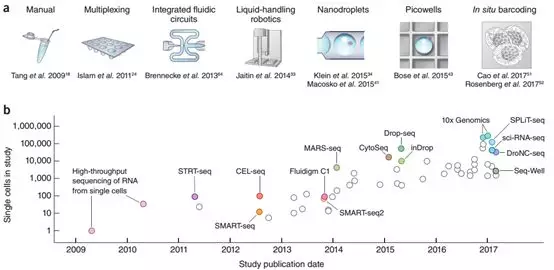
不同于蛋白质,单细胞基因组和转录组已经有广泛的研究。高通量单细胞测序在2009年汤富酬第一篇报道后开始蓬勃发展[2],逐渐形成各式各样的测序方法,检测的细胞数目也有逐年增加的趋势[3]。
单细胞多组学整合也是一大研究趋势,例如染色体开放区域测序(ATAC-seq)与转录组测序数据的整合研究,最近一篇发表在NC上的测序方法scCAT-seq,揭示表观调控的多样性 [4]。单细胞多组学的系统研究有助于进一步剖析细胞不同状态与功能。
前面主要介绍单细胞组学的概况,现在我们开始进入主题,介绍社区划分与模块度的聚类方法。
我们得到单细胞转录组数据以后,经过质控、比对、计算基因表达量、差异基因选择、主成分选取,在PCA的空间中,计算细胞间欧式距离,构建每个细胞的KNN群,最后根据KNN群的交集情况(杰卡德距离)确定细胞与细胞之间权重,得到一个细胞间距离矩阵。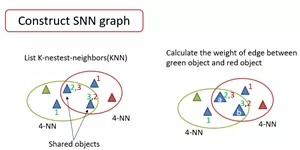
接下来根据细胞之间的权重对细胞群进行分群划分。
聚类的方法有多种。有基于k-mean, 以及k-mean衍生算法,也有基于graph,各式各样。推荐一个github链接,汇总很多方法:https://github.com/seandavi/awesome-single-cell。
本文着重介绍基于graph的社区划分算法。为了方便理解,我把权重01化,如下图所示,9个细胞之间只有有边和无边的关系。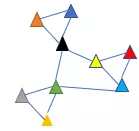
我们基于感官可以判断它们大致可以分为3群,而模块度(modularity)正是赋予了机器这样的判断能力。
模块度取值范围在-1到1之间,数值越大,表示一个分群(group)更加紧凑。一个合理的分群,整体给人的感觉是分群内部的边(蓝色)密度高,而连接外部的边(橙色)比较稀疏。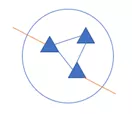
当然,它需要一种量化的方式表示。如果对公式不感兴趣可以跳过这一部分。模块度的公式[5]: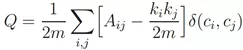
其中: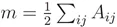
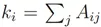
m表示所有细胞之间的权重,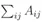 也就是所有边的累加。累加细胞与细胞之间的边,会出现i到j和j到i的重复,所以要除以2。
也就是所有边的累加。累加细胞与细胞之间的边,会出现i到j和j到i的重复,所以要除以2。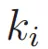 表示连接细胞i的所有边(可能包括连接到分群外部的边(橙色))。
表示连接细胞i的所有边(可能包括连接到分群外部的边(橙色))。
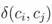 的值只有0和1,当细胞i和j在同一个分群,值为1,相反则为0。所以Q只根据分群内部细胞之间关系累加计算。
的值只有0和1,当细胞i和j在同一个分群,值为1,相反则为0。所以Q只根据分群内部细胞之间关系累加计算。
由公式可以看出,模块度的大小取决于分群内部的边(蓝色)与分群内外部所有边(蓝色橙色)的密度差异。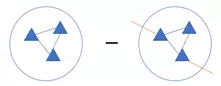
模块度的计算可以简化为:
有了识别分群优劣的能力之后,接下来就可以进行聚类了。我们采用凝聚法聚类,首先将每个细胞识别为一个分群,然后逐一凝聚在一起。每迭代一次凝聚,计算一次模块度,如果凝聚后的模块度比凝聚前的模块度低,那么将放弃本次凝聚结果,继续进行下次凝聚。一直到模块度稳定在最大值。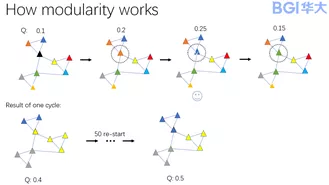
大家有兴趣可以下载脚本自己动手尝试,可以加深理解。
脚本链接:https://github.com/Zhihao-Huang/SingleCellOmics/tree/master/Single_cell_pipeline/test/modularity。
如果分群较多,后续还可以将每个分群作为一个顶点,再进行迭代凝聚。最终得到理想的细胞分群。
基于社区划分的聚类方法适用于细胞数目比较多的情况,如果细胞数少,可能会遗漏掉一些细胞数较少的分群。
参考文献:
[1] Budnik B, Levy E, Harmange G, et al. Mass-spectrometry of single mammalian cells quantifies proteome heterogeneity during celldifferentiation[J]. arXiv preprint arXiv:1808.00598, 2018.
[2] Tang F, Barbacioru C, Wang Y, et al. mRNA-Seq whole-transcriptome analysis of a single cell[J]. Nature methods, 2009, 6(5): 377.
[3] Svensson V, Natarajan K N, Ly L H, et al. Power analysis of single-cell RNA-sequencing experiments[J]. Nature methods, 2017, 14(4): 381.
[4] Liu L, Liu C, Quintero A, et al. Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity[J]. Nature communications,2019, 10(1): 470.
[5] Blondel V D, Guillaume JL, Lambiotte R, et al. Fast unfolding of communities in large networks[J].Journal of statistical mechanics: theory and experiment, 2008, 2008(10): P10008.
如果对内容感兴趣,请关注“单细胞组学”公众号,后续我们会定期推出相关内容,谢谢!

若有收获,就点个赞吧
0 人点赞
