本文来自“单细胞组学”公众号 原文链接:https://mp.weixin.qq.com/s/ewHdefHB33-G-X3i6ex0-A
上期总结了单细胞常用的拟时序算法,本期主要介绍RNA velocity的应用、算法原理和核心代码实现。
内容概要:
- RNA velocity简介与示例
- RNA velocity算法原理
- RNA velocity核心代码实现
- 总结与展望
RNA velocity能做什么?
RNA velocity可以根据单细胞转录组数据,给每个细胞一个显示发展趋势的箭头。
可以显示细胞短时间内的周期变化,如细胞周期[1]。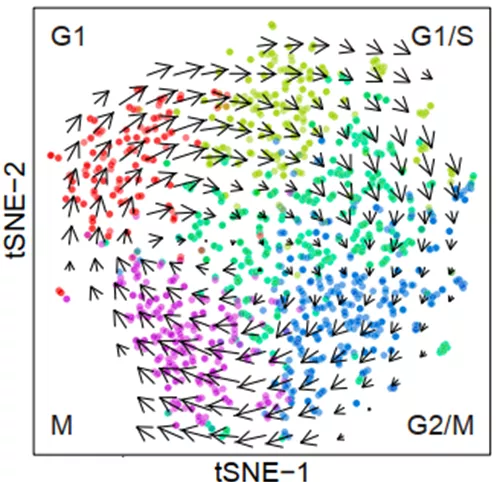
来自MERFISH文章Fig.4 [1]
也可以显示发育轨迹,如海马体单细胞的发育轨迹[2]。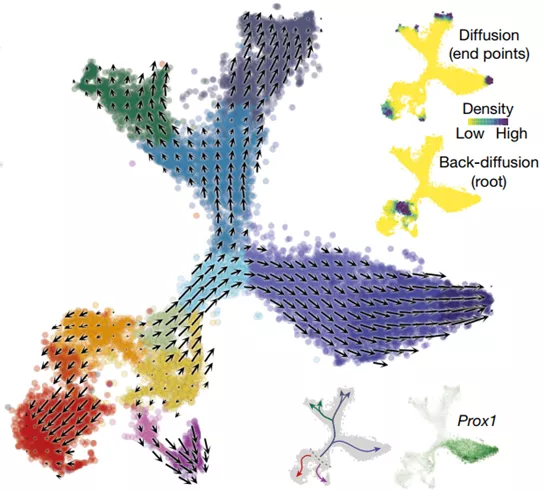
来自RNA velocity文章Fig.3[2]
RNA velocity与其他发育轨迹算法有何不同?
RNA velocity最直观的不同,是它的计算依据,包括两个对象:未剪切mRNA和已剪切mRNA。其他算法大多基于总的mRNA表达矩阵。
RNA velocity生物知识背景
影响单细胞成熟mRNA总量有三个主要的生物进程:
- 转录
- 剪切
- 降解

由于每道工序的运行速率和转换效率存在差异,生物信号的调控,从转录信号发出,到蛋白生成,需要一定时间。这种系统性延迟,可以形成周期性震荡,这对生命体某些功能至关重要,如睡眠周期,心脏跳动的脉冲信号等。
我们用α,β,γ分别表示转录,剪切,降解这三个过程,用u和s分别表示未剪切mRNA的量和已剪切mRNA的量。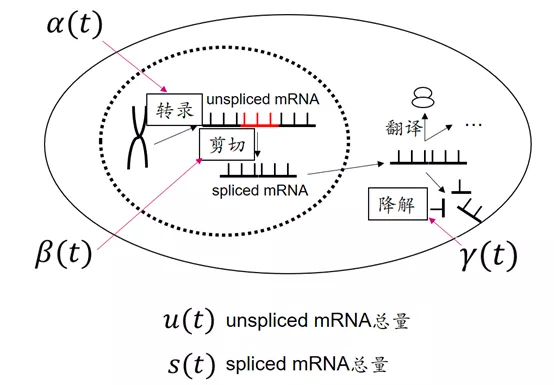
某个基因的调控过程,可以表现为:转录起始信号α发出,未剪切mRNA的量u逐渐增多,紧随着,已剪切mRNA的量s也开始逐渐增多,如下右图。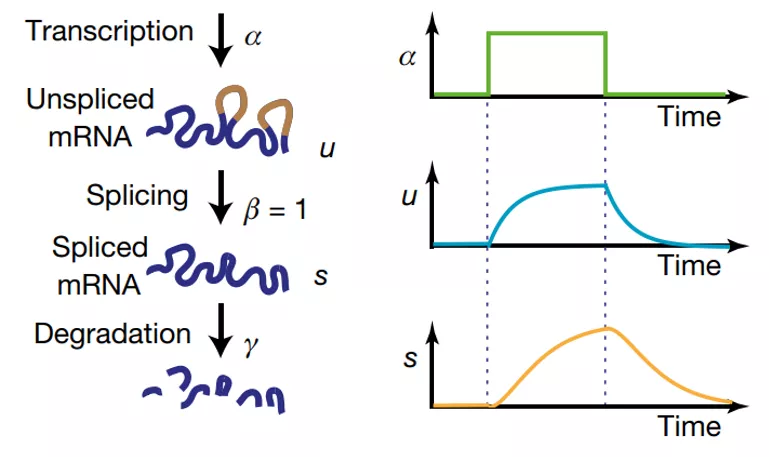
这种调控模式类似系统生物学中经典的前馈网络,共门I型前馈网络( coherent type-1 feed-forwardloop, C1-FFL),如下图所示。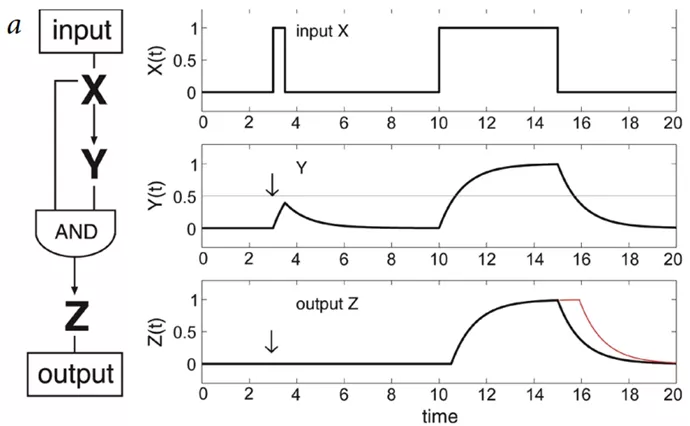
I型前馈网络+共门。Y基因只有达到一定阈值时,才能促发Z基因的表达。意义在于,过滤噪声刺激,降低敏感性[2]。
RNA velocity模型
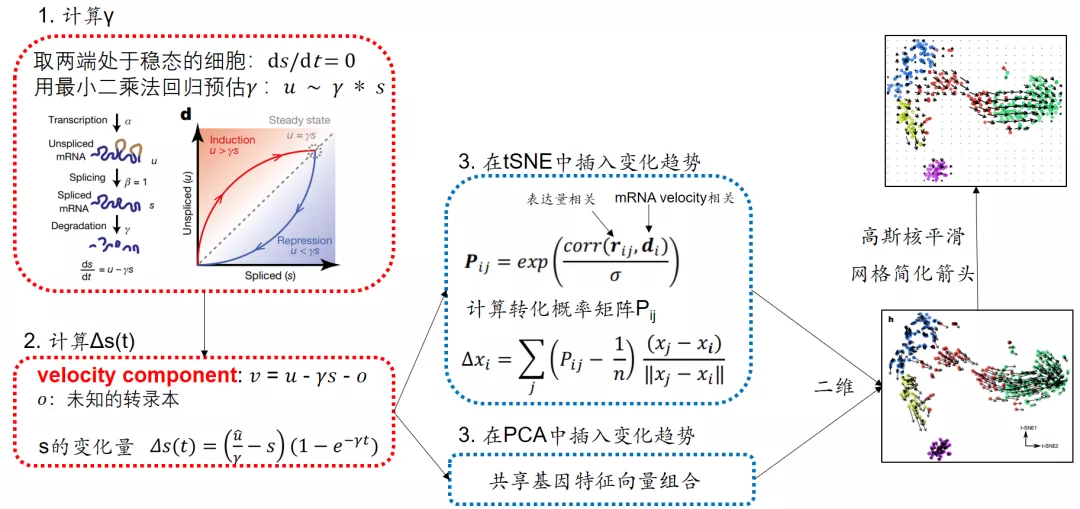
为了得到每个细胞的发展趋势,RNA velocity首先计算每个基因已剪切mRNA总量s的变化速率,然后算出未来某一时间t的已剪切mRNA总量s(t), 最后根据线性或者非线性的方法,将s(0)和s(t)插到二维图中,代表箭头的两端,表示每个细胞的发展趋势。
简要步骤:
- 计算每个基因的降解常数γ;
- 计算每个基因的s(t);
- 在PCA或者tSNE中可视化趋势。
1. 预估降解常数γ
根据上文提到的5个对象进行建模,未剪切mRNA的量u和已剪切mRNA的量s的变化速率可以表示为: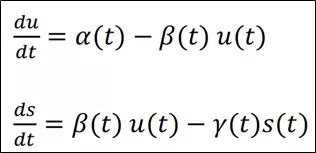
可以对模型进行简化。假设转录速率α不变,剪切效率完美,β=1,最终得到𝑢和γ*𝑠的关系: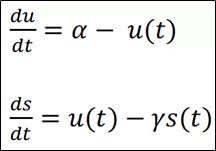
已剪切mRNA总量s的变化速率= 未剪切mRNA总量 u–降解系数γ x s
根据scRNA-seq数据可以到u和s表达矩阵。在s稳定的条件下,即ds/dt= 0时,u = γs,我们就可以根据线性回归的方法,预估每个基因的γ值。
如何确定s恒定不变的条件呢?
下图用另一种方式展示上文的前馈网络:首先转录,纵坐标u逐渐增多,圆点开始往上升,随着剪切过程的进行,横坐标s也增多,圆点开始往右边移动。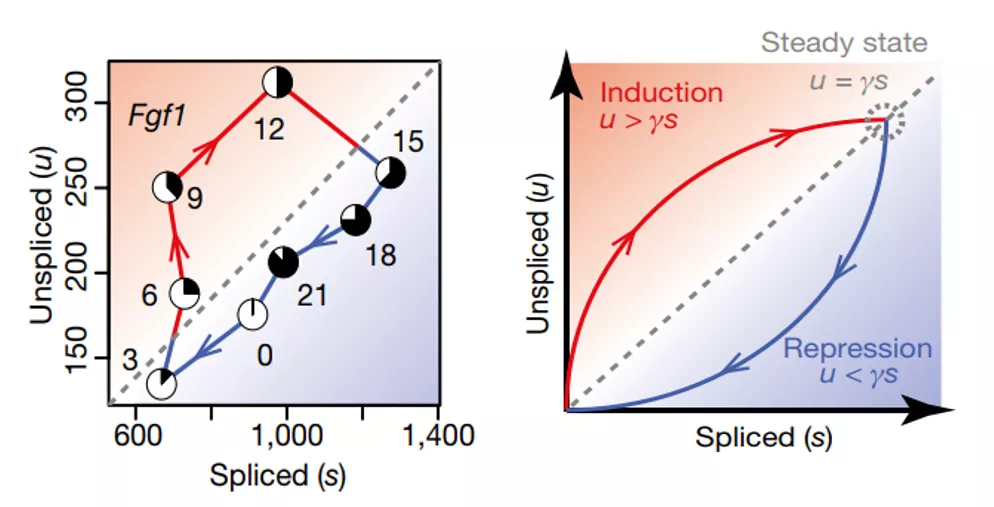
当已剪切mRNA总量s处于稳态时,生成s的量和降解s的量相等,u达到最大值,也就是上图右图虚线表示的区域。这是递增后的稳态。相反的,也存在递减后的稳态。
因此,满足s恒定条件的细胞,处于u取值区间的两端。我们可以根据这些满足条件的细胞,回归计算γ值。
2. 预估s(0)未来的变化趋势s(t)
求解上述的微分方程:

最终得到: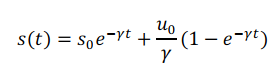

因此,根据时间=0时的三个数值:未剪切mRNA总量u0,已剪切mRNA总量s0,γ值,代入公式可以计算时间=t时的已剪切mRNA总量s(t)。
考虑并非所有的转录本都有注释,非剪切mRNA总量u里有部分是未知的,我们把这些干扰定义为o。通过s非0且u=0的细胞比例量化o。最终的s变化速率: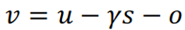
经过o调整后的未剪切mRNA总量: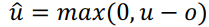
最后得到经过t时间s的变化量Δs(t) = s(t) – s0: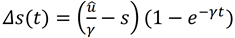

3. 在PCA中可视化
根据s0的基因特征向量组合,计算s(t)细胞特征向量组合。如果细胞过多,箭头过于密集,可以用高斯核平滑的方法,网格化箭头。
4. 在tSNE中可视化
非线性降维的插入比较复杂,首先根据一个细胞的表达量与其他细胞的RNA velocity向量的相关系数,经过指数核函数变换,得到转移概率矩阵P。

然后累加与其他细胞的P与cosine距离的相乘,得到箭头向量:
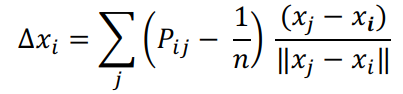
核心代码实现
测试数据选取MERFISH的数据,来自MERFISH研究细胞周期的文章[1]。
1. 测试数据导入
#load dataemat <- read.table('emat.txt')nmat <- read.table('nmat.txt')comdf <- read.table('cell_cluster.txt', header = T)cell.colors <- comdf$colornames(cell.colors) <- comdf$cellID
- 分别标准化矩阵s和u。
参数解读:
mult:测序深度,也就是标准化的scale factor; ```r
rvel.cd.unpooled <- list();
bring matrices to the same gene set (just in case)
vg <- intersect(rownames(emat),rownames(nmat)); emat <- emat[vg,]; nmat <- nmat[vg,] pcount <- 1;
size estimates
emat.size <- Matrix::colSums(emat) nmat.size <- Matrix::colSums(nmat) emat.cs <- emat.size[colnames(emat)]/mult; nmat.cs <- nmat.size[colnames(nmat)]/mult; emat.log.norm <- log(as.matrix(t(t(emat)/emat.cs))+pcount);
size-normalized counts
conv.emat.norm <- t(t(emat)/emat.cs) conv.nmat.norm <- t(t(nmat)/nmat.cs) rvel.cd.unpooled$conv.nmat.norm <- conv.nmat.norm; rvel.cd.unpooled$conv.emat.norm <- conv.emat.norm;
3. 回归计算γ值<br />参数解读:<br />fit.quantile:定义稳态细胞的两端表达区间的比例;根据数据而定,Smart-Seq 5%, droplet数据2%;<br />steady.state.cells:自定义稳态细胞;<br />deltaT、deltaT2:时间变化单位,默认为1.<br />min.nmat.emat.correlation:根据u和s表达量的相关系数过滤基因;<br />min.nmat.emat.slope:根据γ值过滤基因;```rko <- data.frame(do.call(rbind,parallel::mclapply(sn(rownames(conv.emat.norm)),function(gn) {df <- data.frame(n=(conv.nmat.norm[gn,steady.state.cells]),e=(conv.emat.norm[gn,steady.state.cells]))o <- 0;zi <- df$e<1/emat.cs[steady.state.cells];if(any(zi)) { o <- sum(df$n[zi])/(sum(zi)+1)}df$o <- o;eq <- quantile(df$e,p=c(fit.quantile,1-fit.quantile))pw <- as.numeric(df$e>=eq[2] | df$e<=eq[1])d <- lm(n~e,data=df,weights=pw)# note: re-estimating offset herereturn(c(o=as.numeric(coef(d)[1]),g=as.numeric(coef(d)[2]),r=cor(df$e,df$n,method='spearman')))},mc.cores=n.cores,mc.preschedule=T)))ko <- na.omit(ko)vi <- ko$r>min.nmat.emat.correlationko <- ko[vi,]vi <- ko$g>min.nmat.emat.slopeko <- ko[vi,]
- 计算s(t),即projected s。
gamma <- ko$g; offset <- ko$o; names(gamma) <- names(offset) <- rownames(ko); cat("calculating RNA velocity shift ... ") deltaE <- t.get.projected.delta(conv.emat.norm,conv.nmat.norm,gamma,offset=offset,delta=deltaT) rvel.cd.unpooled$gamma <- gamma; # reduced cell normalization (only genes for which momentum was estimated) emat.norm <- emat[rownames(emat) %in% rownames(deltaE),] emat.sz <- emat.cs; emat.norm <- t(t(emat.norm)/(emat.sz)); emn <- t.get.projected.cell2(emat.norm,emat.sz,as.matrix(deltaE),mult = mult,delta=deltaT2); cat("done\n") full.ko$valid <- rownames(full.ko) %in% rownames(ko) rvel.cd.unpooled <- c(rvel.cd.unpooled,list(projected=emn,current=emat.norm,deltaE=deltaE,deltaT=deltaT,ko=full.ko,mult=mult,kCells=1)); - 在PCA中可视化趋势
pca.velocity.plot.simplify(rvel.cd.unpooled, nPcs=2, plot.cols=1, cell.colors=cell.colors, pc.multipliers=c(1,-1) ## adjust as needed to orient pcs )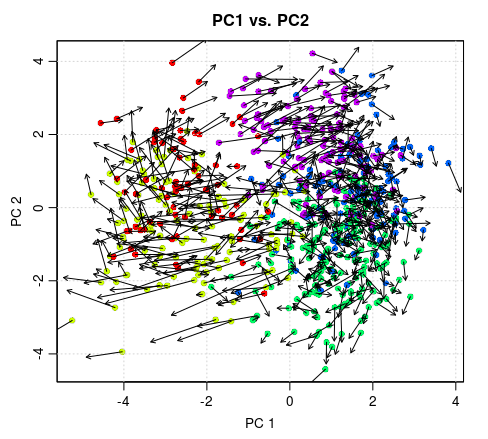
网格化pca.velocity.plot.simplify(rvel.cd.unpooled, nPcs=2, plot.cols=1, cell.colors=cell.colors, pc.multipliers=c(1,-1), ##adjust as needed to orient pcs show.grid.flow = TRUE, grid.n=20 ## adjust as needed )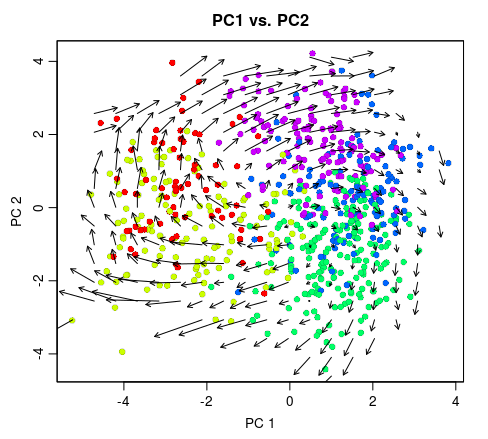
6. 显示细胞周期###plot cell-cycle phase library(Seurat) library(ggplot2) rownames(comdf) <- comdf$cellID seus4 <- CreateSeuratObject(counts =emat, meta.data = comdf) seus4 <- CellCycleScoring(object =seus4, g2m.features = cc.genes$g2m.genes, s.features =cc.genes$s.genes) #pca x0 <- rvel.cd.unpooled$current x1 <- rvel.cd.unpooled$projected message("Step1. log ... ") x0.log <- log2(x0 + 1) x1.log <- log2(x1 + 1) message("Step2. pca ... ") cent <- rowMeans(x0.log) epc <- pcaMethods::pca(t(x0.log - cent),center = F, nPcs = 2) message("Step3. pc multipliers ...") epc@loadings <- t(t(epc@loadings) *c(1,-1)) epc@scores <- scale(epc@completeObs,scale = F, center = T) %*% epc@loadings head(epc@scores) ccdf <- as.data.frame(epc@scores) ccdf <- cbind(ccdf,seus4@meta.data[,c('S.Score','G2M.Score','Phase')]) ggplot(ccdf, aes(PC1, PC2, color = Phase))+ geom_point() + theme_bw()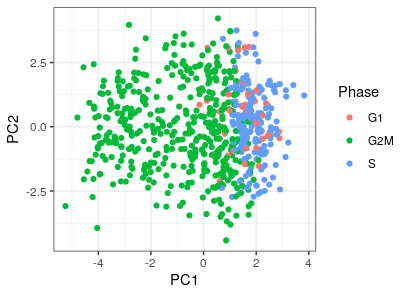

显示细胞周期S和G2M的分数ggplot(ccdf, aes(PC1, PC2, color =S.Score)) + geom_point() + theme_bw() + scale_color_gradientn(colours = c('grey','yellow','red')) ggplot(ccdf, aes(PC1, PC2, color =G2M.Score)) + geom_point() + theme_bw() + scale_color_gradientn(colours = c('grey','yellow','red'))
详细源码和示例数据可以查看github链接:
https://github.com/Zhihao-Huang/SingleCellOmics/blob/master/000.RNA_velocity_simplify.R总结与展望
RNA velocity原理可以分为两大部分:Δs(t)计算和可视化。总结为下图。
2. RNA velocity最大的不足之处是稳态细胞的定义。2020年新出scVelo用新的算法弥补这一缺陷[4]。举一反三,将RNA velocity推广至单细胞多组学,如“蛋白质 velocity”[5]。
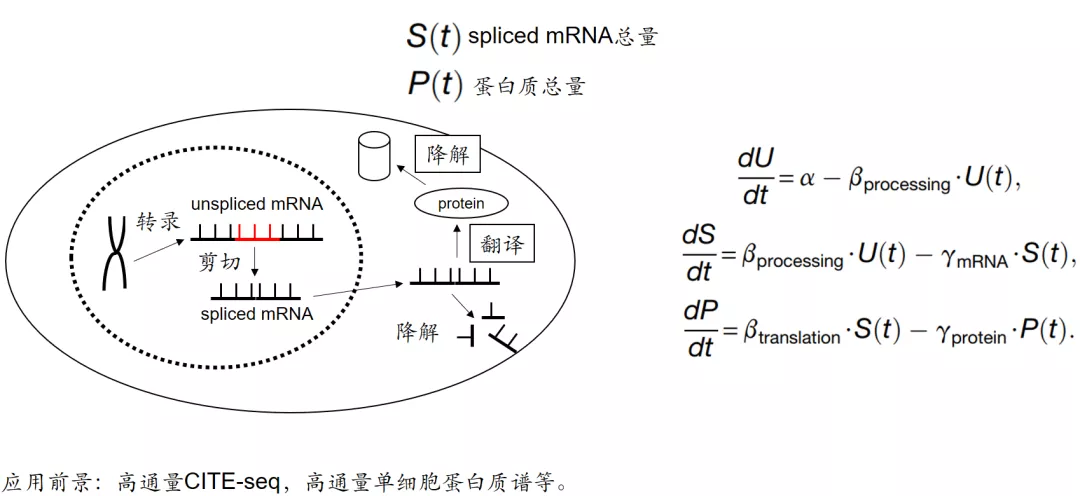
“ATAC velocity” 类似的思路也有人探索,如Share-seq这一类可以检测同一个细胞的染色体开放区域和转录组的技术[6]。随着单细胞多组学技术逐渐完善,迫切需要寻找不同组学之间的共同规律,使之成为共同体。RNA velocity这一类的分子动力学模型,将是多组学分析的有力工具之一。
参考链接:
[1] Xia C, Fan J, Emanuel G, et al. Spatial transcriptomeprofiling by MERFISH reveals subcellular RNA compartmentalization and cellcycle-dependent gene expression[J]. Proceedings of the National Academy of Sciences, 2019, 116(39): 19490-19499.
[2] La Manno G, Soldatov R, Zeisel A, et al. RNA velocity ofsingle cells[J]. Nature, 2018, 560(7719): 494-498.
[3] Shen-Orr S S, Milo R, Mangan S, et al.Network motifs in the transcriptional regulation network of Escherichiacoli[J]. Nature genetics, 2002, 31(1): 64-68.
[4] Bergen V, Lange M, Peidli S, et al. Generalizing RNA velocity to transient cell states through dynamical modeling[J]. Nature biotechnology, 2020, 38(12): 1408-1414.
[5] Gorin G, Svensson V, Pachter L. Protein velocity and acceleration from single-cell multiomics experiments[J]. Genome biology, 2020, 21(1): 1-6.
[6] Clyde D. SHARE-seq reveals chromatin potential[J]. Nature Reviews Genetics, 2020: 1-1.

若有收获,就点个赞吧
0 人点赞
