本文来自“单细胞组学”公众号 原文链接:https://mp.weixin.qq.com/s/rOCOH5SAtkWf0WkDditIPw
内容概要
1. 10x Visium空间转录组技术介绍
2. 10x Visium空间转录组分析流程,以Seurat为例
3. 单细胞转录组整合分析
4. 解锁空间聚类新方法:基因共表达与空间位置关联分析(scHOT)
5. 空间转录组位点细胞比例分解方法:
- MIA
- Giotto
- SPOTlight
- bisqueRNA
- CIBERSORTx
- 空间转录组分析新思路与方法
空间基因检测技术汇总
近几年来,空间基因检测技术开始蓬勃发展。目前以RNA/蛋白质为检测对象的技术有下图展示的9种[1]。圆点表示检测对象是RNA,三角形表示蛋白。不同颜色代表不同的分辨率,红色代表比较精确的单细胞水平。可以从图中看出,单细胞水平的多基因检测技术只有seqFISH+,而目前市场上10x Visium的检测单位是直径55微米的斑点(spot)。

10x Visium空间转录组技术由SPATIAL TRANSCRIPTOMICS发展而来。SPATIAL TRANSCRIPTOMICS公司由Joakim Lundeberg等人于2016年在瑞典斯德哥尔摩创立。在同一年Joakim Lundeberg等人在Science发表空间转录组文章。公司在2018年被10x Genomics收购,成为其旗下子公司。

起初Spatial transcriptomic 检测精度不高,每个spot的直径100微米,spot间的距离200微米。10x Genomics后续对该空间转录组技术进行改进,于是有了现在的10x Visium,每个spot直径55微米,spot间距100微米。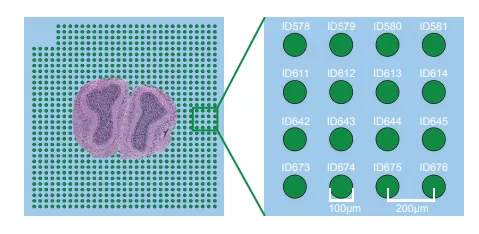

一张visium空间基因表达玻片上有4个边长为0.65cm的正方形区域,可以检测4张组织切片。每个捕获区域有5000个spot,每个spot相当于单细胞转录组建库中用于结合单个细胞的磁珠,类似于同个磁珠上有相同的细胞barcode,同个spot上也有统一的spatial barcode。这个spatial barcode可以用于后续分析的spot空间坐标定位[2] 。
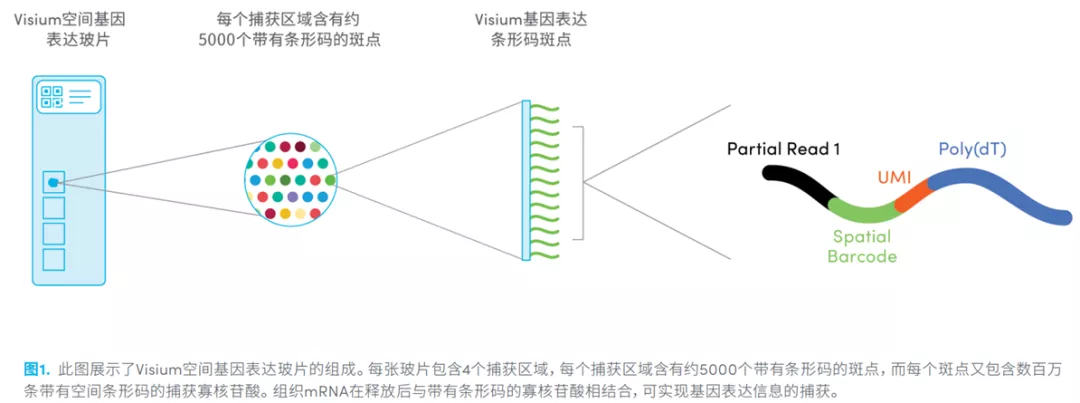
空间转录组分析流程
分析流程跟单细胞转录组分析流程类似。不一样的地方主要有两点:
1. 空间转录组免疫组化图片和spot的组织坐标信息;
2. spot是组织的一个小区域,不一定是单个细胞,所以在聚类后分群鉴定时,不能直接用细胞类型特异的marker基因确定细胞类型,可能存在多种细胞类型在同一个spot里。
所以空间转录组往往需要组织相对应的单细胞转录组的细胞类型特征做辅助鉴定。如单细胞转录组整合分析,或者基于单细胞转录组特征基因做deconvolution,获取每个spot的细胞类型比例。
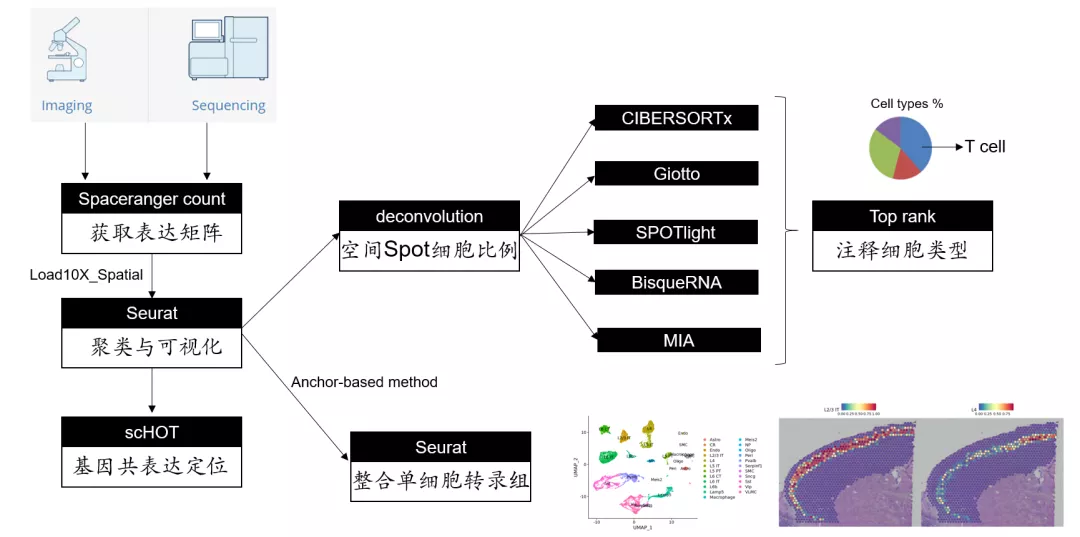
Seurat分析流程结果解读
单细胞转录组分析R包Seurat已经有10x Visium空间转录组的分析流程。过程跟单细胞转录组分析流程类似。大家可以上Seurat官网查看[3]。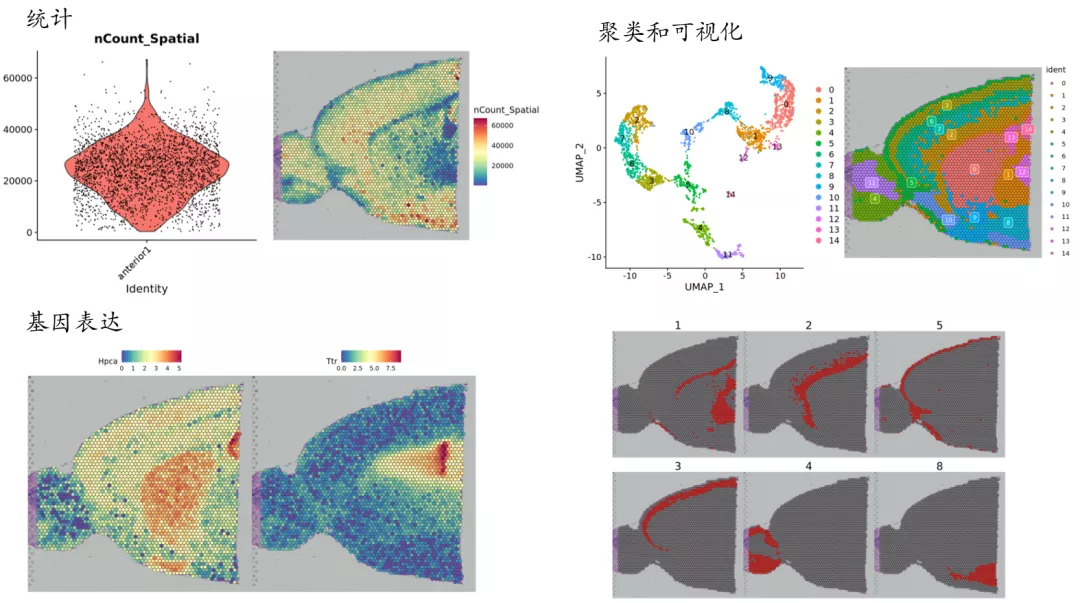
整合单细胞转录组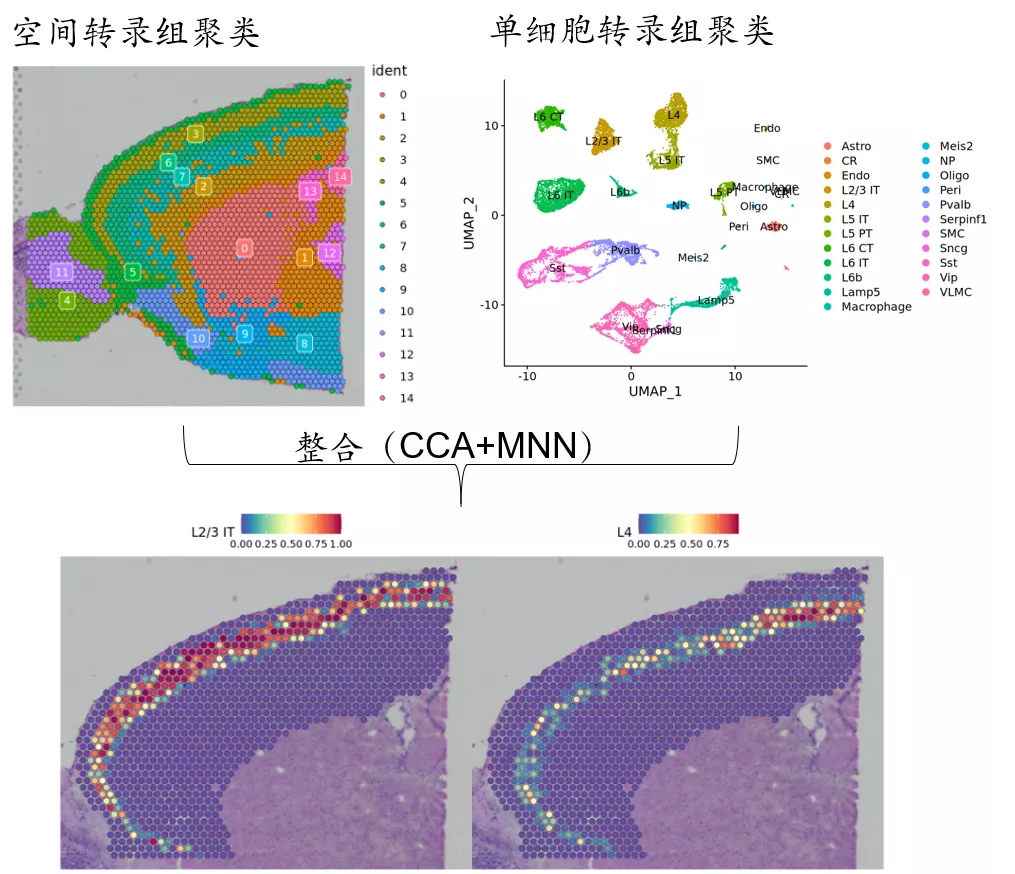
解析bulk样本细胞类型比例的一般方法(deconvolution)
鉴定空间转录组细胞类型,除了用整合单细胞转录组的方法之外,还可以用deconvolution的方法。一般只需要提供空间转录组表达矩阵和单细胞数据的细胞类型特征基因矩阵就可以了。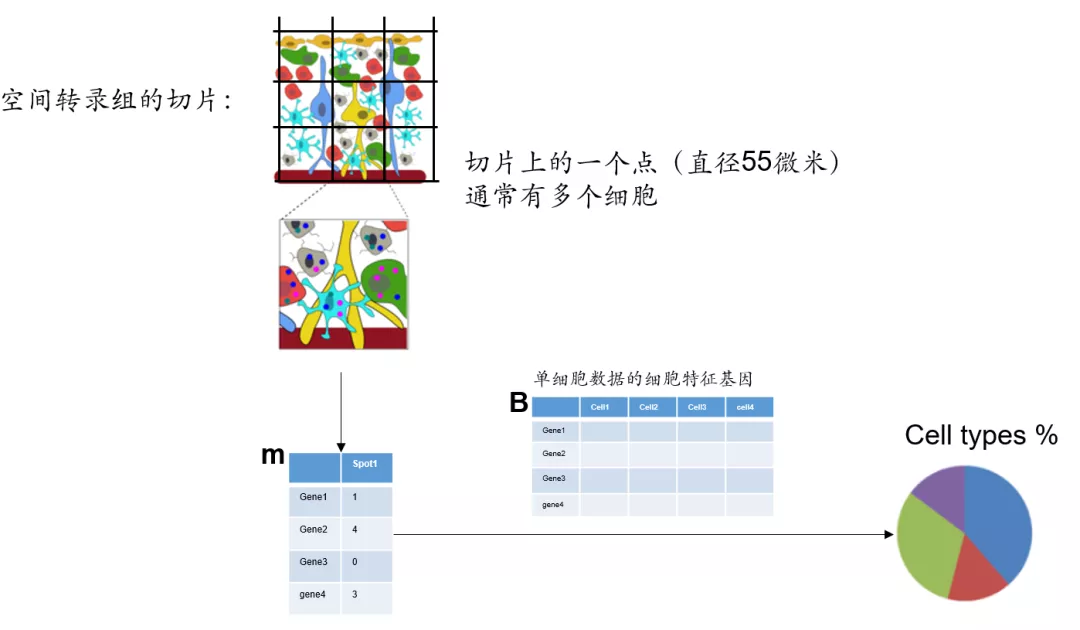
细胞类型的Deconvolution如何工作?
最基本的细胞类型deconvolution方法是基于线性的方法,其基本假设是spot每个基因的表达量,由spot中每个细胞该基因表达量的累加。基于该假设,就可以计算出每个spot的细胞类型比例,以及每个细胞类型的表达量。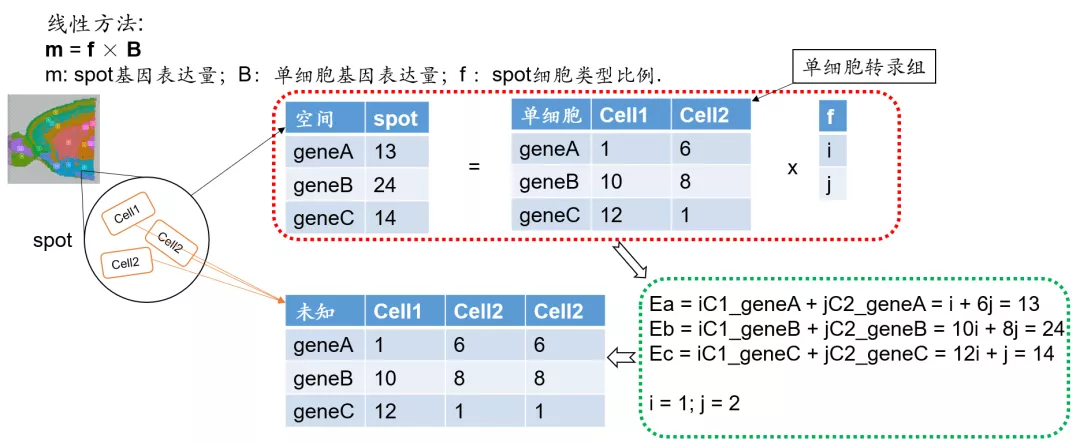
线性方法的缺陷:
1. 不完整细胞产生误差;
2. 对噪声敏感。
基础的线性方法存在缺陷,于是有人开发基于多细胞deconvolution的方法。
5种空间转录组deconvolution的方法
下图是我整理并测试过的5种deconvolution方法。其中MIA和Giotto是基于空间转录组设计的方法,而bisqueRNA/SPOTlight/CIBERSORTx的设计初衷是解决bulk样本的细胞组分问题。所有方法都有一个相同的依赖,就是单细胞转录组的细胞类型特征基因。
1. multimodal intersection analysis (MIA)

MIA是今年年初NBT一篇空间转录组文章展示的方法[4]。文章的经典设计套路值得学习。一个肿瘤部位同时做空间转录组和单细胞转录组,最后用单细胞转录组注释空间转录组。虽然空间转录组还是用SPATIAL TRANSCRIPTOMIC的老技术,但是也不妨碍他们发NBT。
MIA原理
MIA与GO富集的思路如出一辙。将单细胞的特征基因作为GO terms,用空间转录组无监督聚类分群的差异基因做富集即可。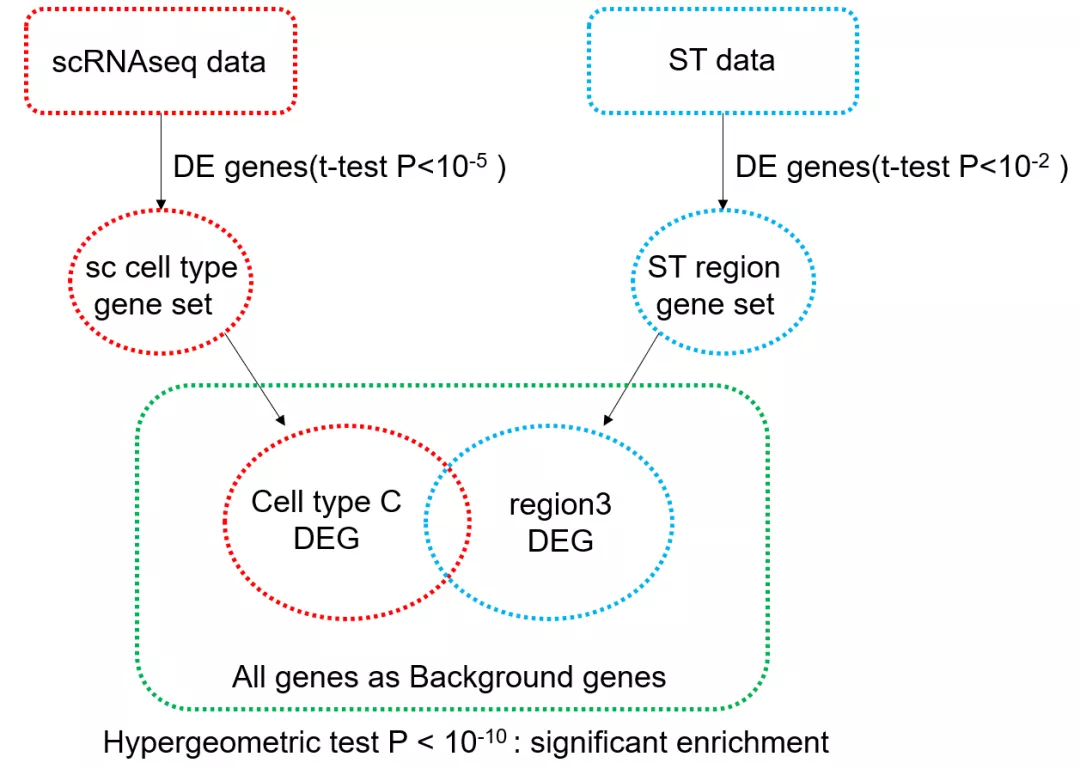
MIA用于解析空间转录组细胞比例
下图是NBT这篇文章展示胰腺癌切片。最后的细胞类型富集结果用右边的热图表示。**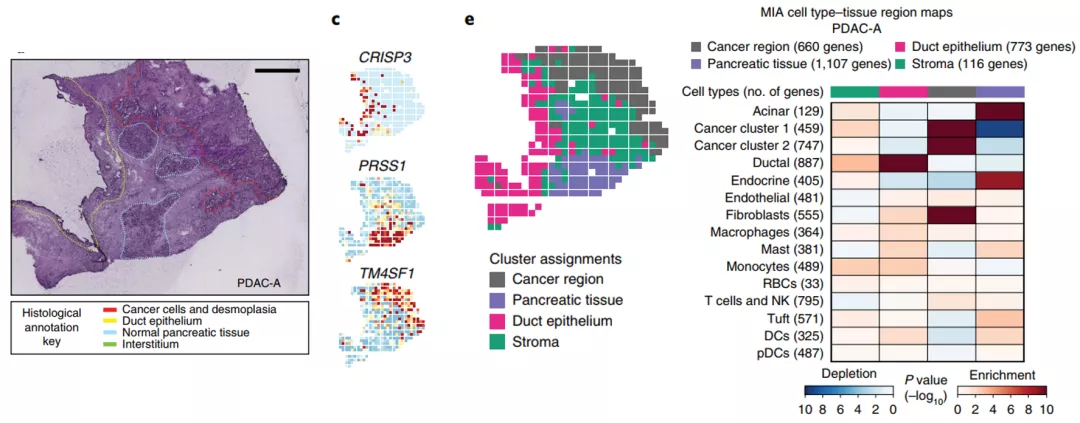
2. Giotto(method based on enrichment)
Giotto类似于MIA也是用富集的方法[1],但富集方法选择多一些。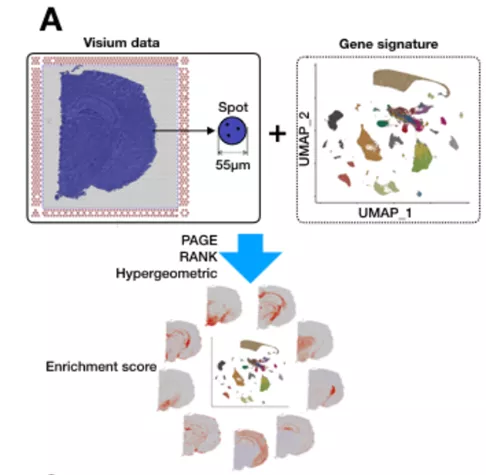
步骤:
1.获取单细胞转录组的每个细胞类型的特征基因集;2.选择一种富集方法:
PAGE(GSEA);
RANK;
Hypergeometric(GO);
3. 以spot为对象,特征基因集为参考,做富集分析;
4. 最后每个spot可以得到细胞类型比例。
3. SPOTlight (method based on NMF)
SPOTlight是基于非负矩阵分解(NMF)和非负最小二乘法(NNLS)做的deconvolution[5]。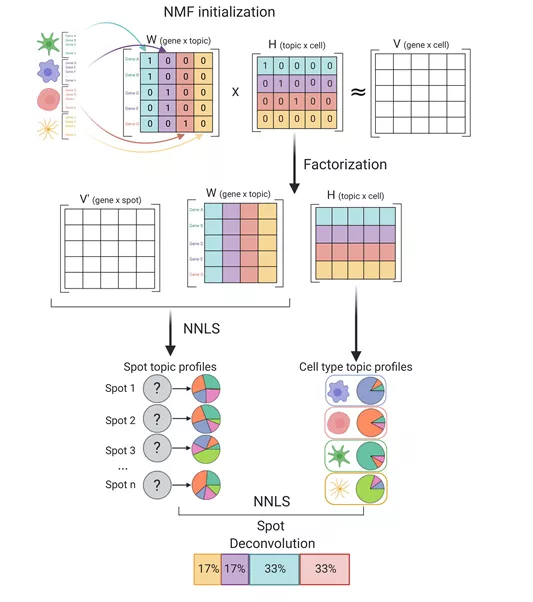
主要步骤:
1. 矩阵V是单细胞转录组的特征基因矩阵;对V做非负矩阵分解,得到W和H。非负矩阵分解的topic个数设置为细胞类型的个数。
2. 取W矩阵(genex topic)和空间转录组的表达矩阵V’做非负最小二乘(NNLS),得到Spot和topic的矩阵;
3. Spotxtopic的矩阵与之前的H矩阵(topicx cell)做非负最小二乘,得到Spot 和细胞比例的矩阵。
用SPOTlight解析Seurat流程用的小鼠大脑示例数据[6]: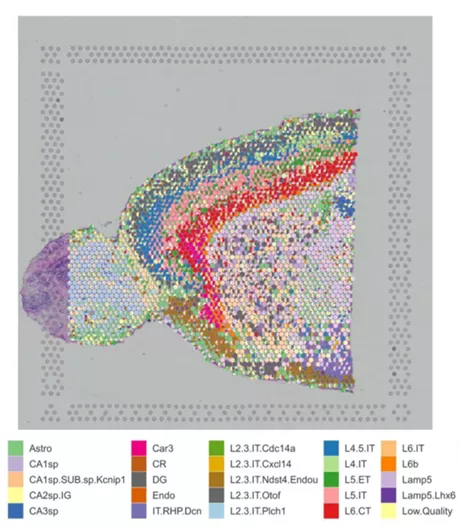
4. bisqueRNA
BisqueRNA是一种基于回归的方法[7]。首先,需要生成单细胞特征矩阵;然后关联bulk转录组的分布,解析spot比例。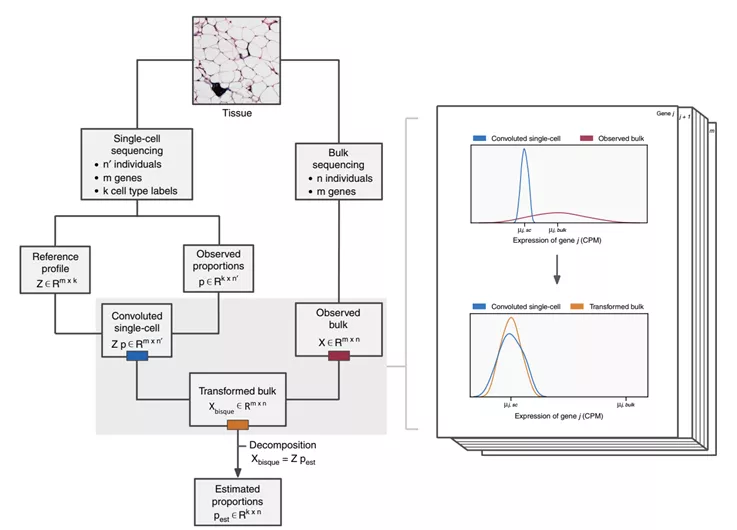
5. CIBERSORTx
CIBERSORTx是由CIBERSORT发展而来[6]。
优点:
1. 操作方便,提供网站在线分析。用教育网邮箱注册即可使用;
2. 单细胞特征基因自动寻找,无须花时间设置参数;
3. 提供一定程度的可视化;
4. 结果对噪声不敏感,纠正线性损失函数造成的过拟合。
6. scHOT:根据基因表达相关性确定不同空间模块[8]
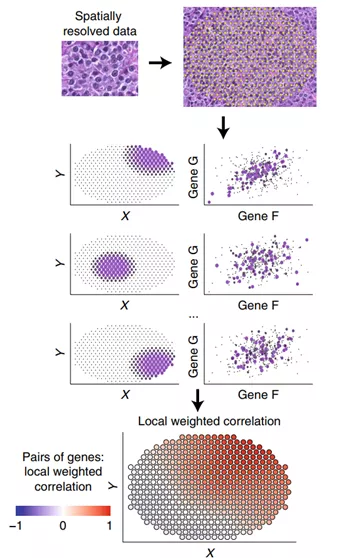

空间转录组分析思路和想法
可以开发的新方法:
1. 结合HIC图像的聚类(跟染色剂相关);
2. 结合空间周围细胞类型的比例分解;
之前介绍几种deconvolution的方法都是将单独spot为研究对象,没有考虑spot的位置。
3. 更高的解析度:单细胞/亚细胞,多基因。
seqFISH+:单个细胞可以检测10000个基因的表达量[9]
每个通道20种伪颜色,三轮203=8000种组合编码;三个通道加起来理论上可以检测24000个基因。
Giotto应用于seqFISH+数据的细胞比例解析
1.切片分区;
2.以每个小网格为对象做细胞类型富集分析,类似于10x Visium的spot。
参考资料:
[1]. Dries, Ruben, et al. “Giotto, a pipeline for integrative analysis and visualization of single-cell spatial transcriptomic data.” BioRxiv (2019): 701680.
[2].https://pages.10xgenomics.com/rs/446-PBO-704/images/CN_10x_BR060_Inside_Visium_Spatial_Technology_A4_Digital.pdf
[3]. https://satijalab.org/seurat/v3.2/pbmc3k_tutorial.html
[4]. Moncada R, Barkley D, Wagner F, et al.Integrating microarray-based spatial transcriptomics andsingle-cell RNA-seqreveals tissue architecture in pancreatic ductal adenocarcinomas[J]. NatureBiotechnology, 2020, 38(3): 333-342.
[5]. Elosua, Marc, et al. “SPOTlight: Seeded NMF regression to Deconvolute Spatial Transcriptomics Spots with Single-Cell Transcriptomes.” BioRxiv (2020).
[6]. JewB, Alvarez M, Rahmani E, et al. Accurate estimation ofcell composition in bulk expression through robust integration of single-cellinformation[J]. Nature Communications, 2020, 11(1): 1-11.https://doi.org/10.1038/s41467-020-15816-6
[7]. JewB, Alvarez M, Rahmani E, et al. Accurate estimation ofcell composition in bulk expression through robust integration of single-cellinformation[J]. Nature Communications, 2020, 11(1): 1-11.
[8]. Ghazanfar S, Lin Y, Su X, et al.Investigating higher-order interactions in single-cell data with scHOT[J]. Nature Methods, 2020: 1-8.
[9]. Eng, Chee-Huat Linus, et al. “Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+.” Nature 568.7751 (2019): 235-239.

若有收获,就点个赞吧
0 人点赞
