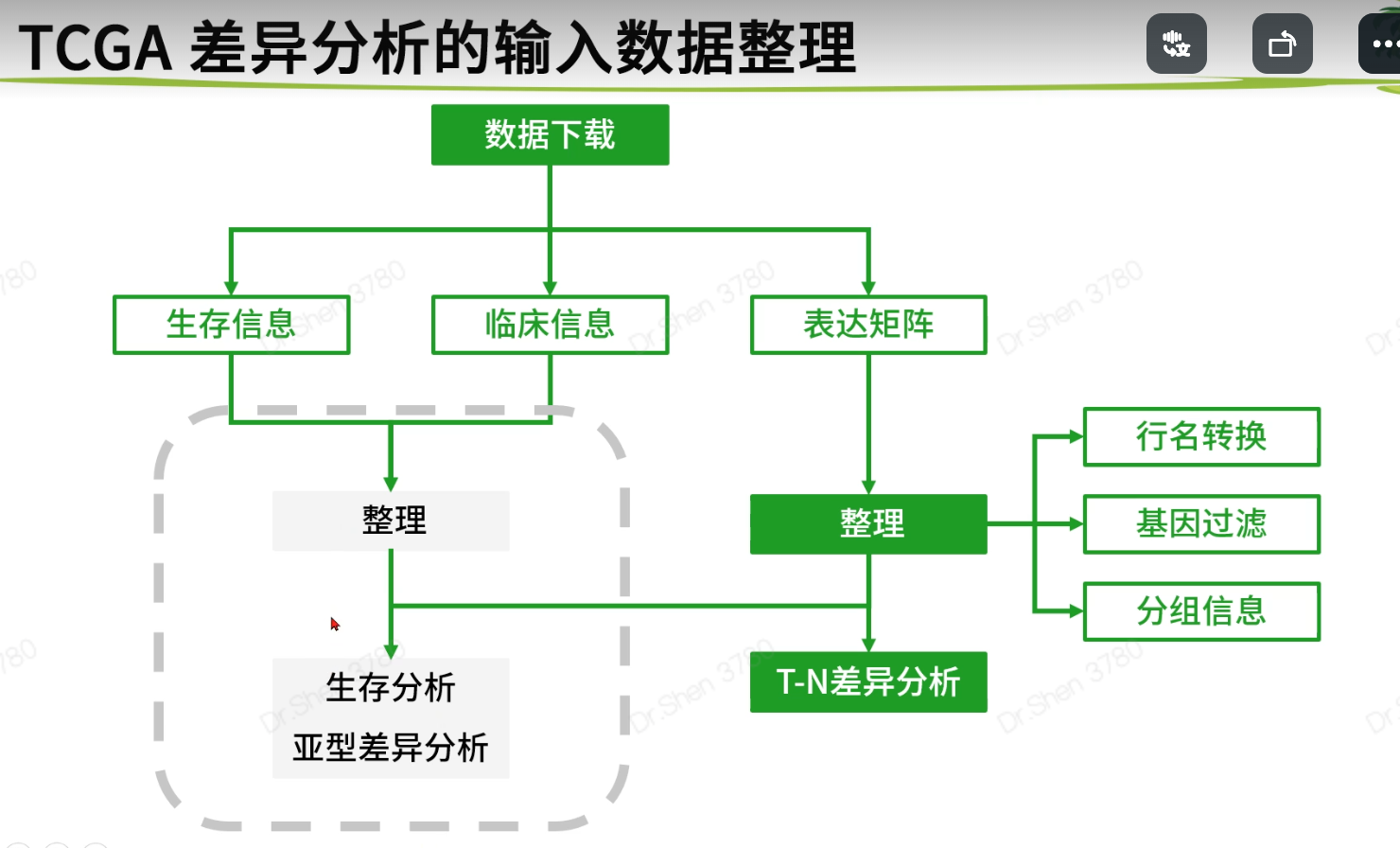
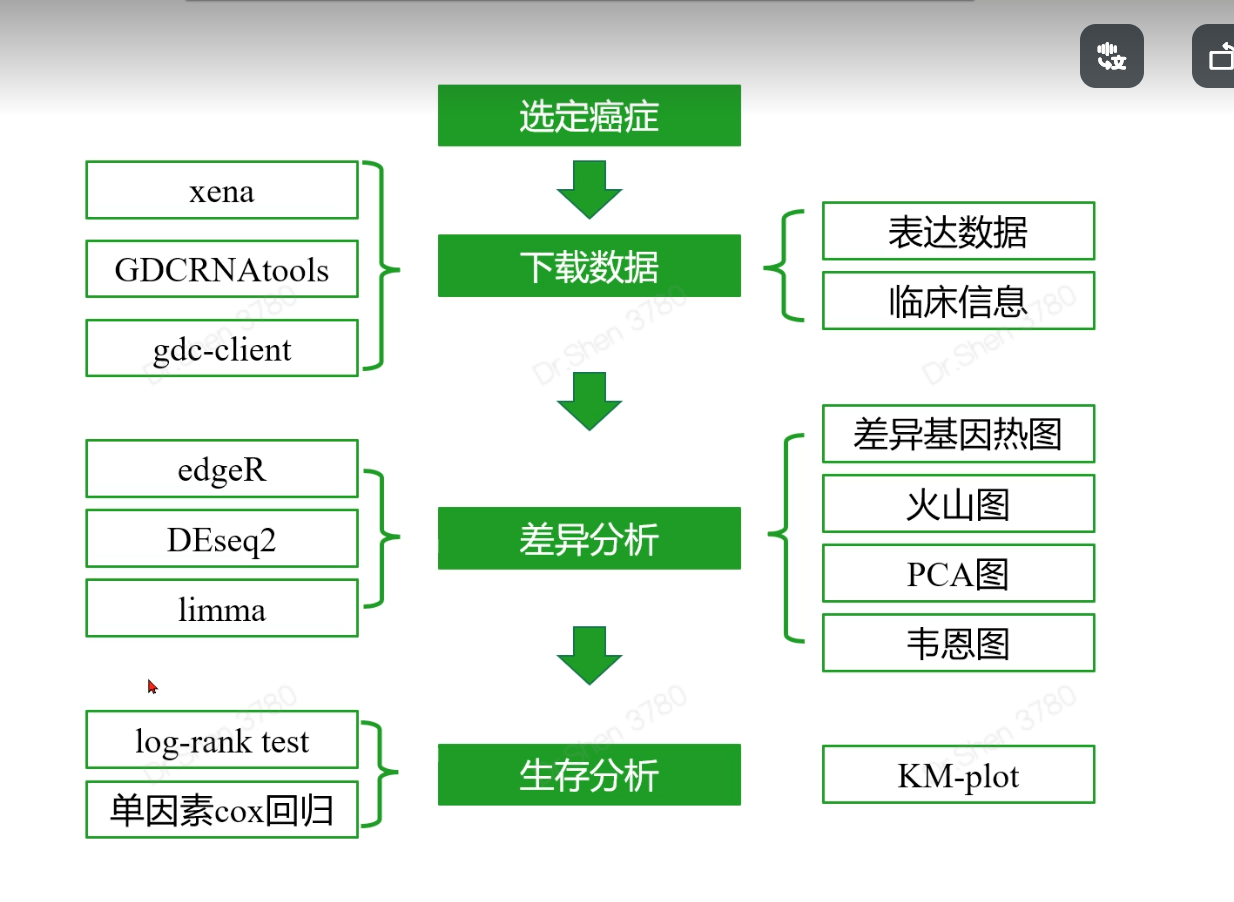
前期介绍
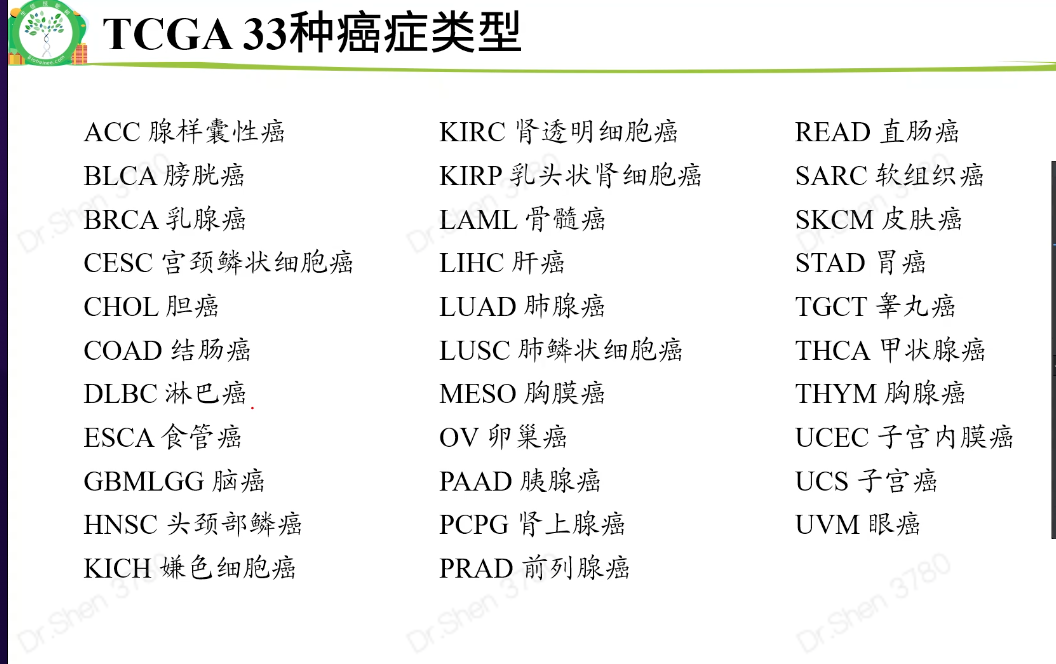
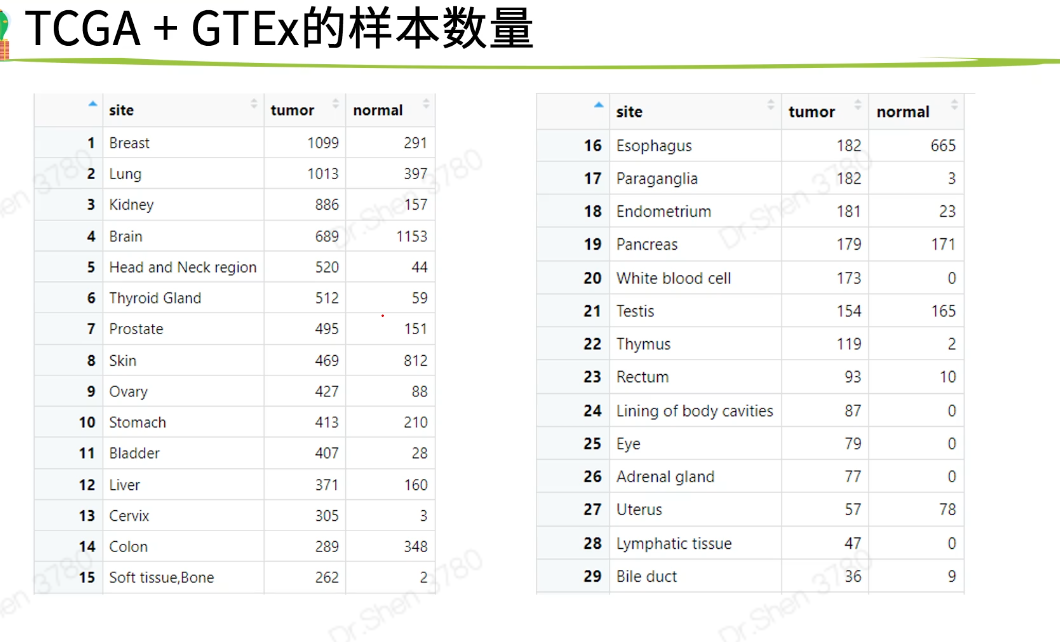
癌症类型
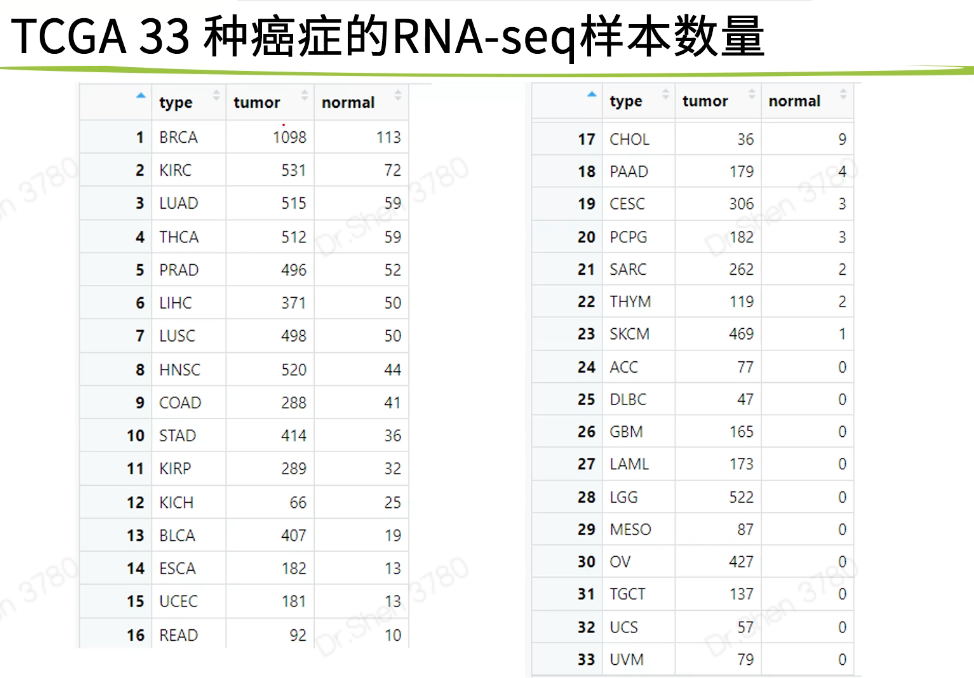
全部对比或配对样本;样本数量少时差可从别的数据中进行差异?生存?等后续分析
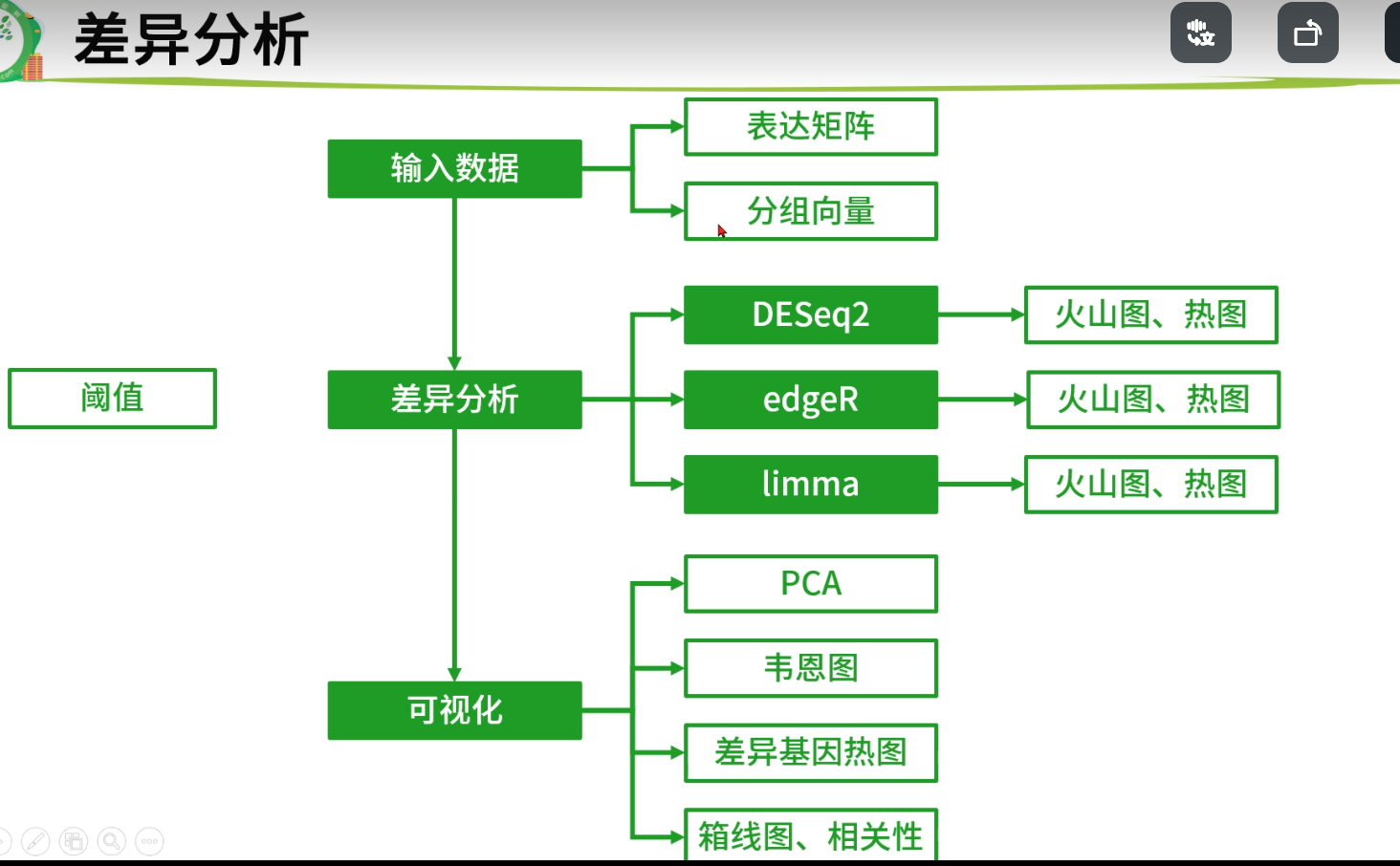
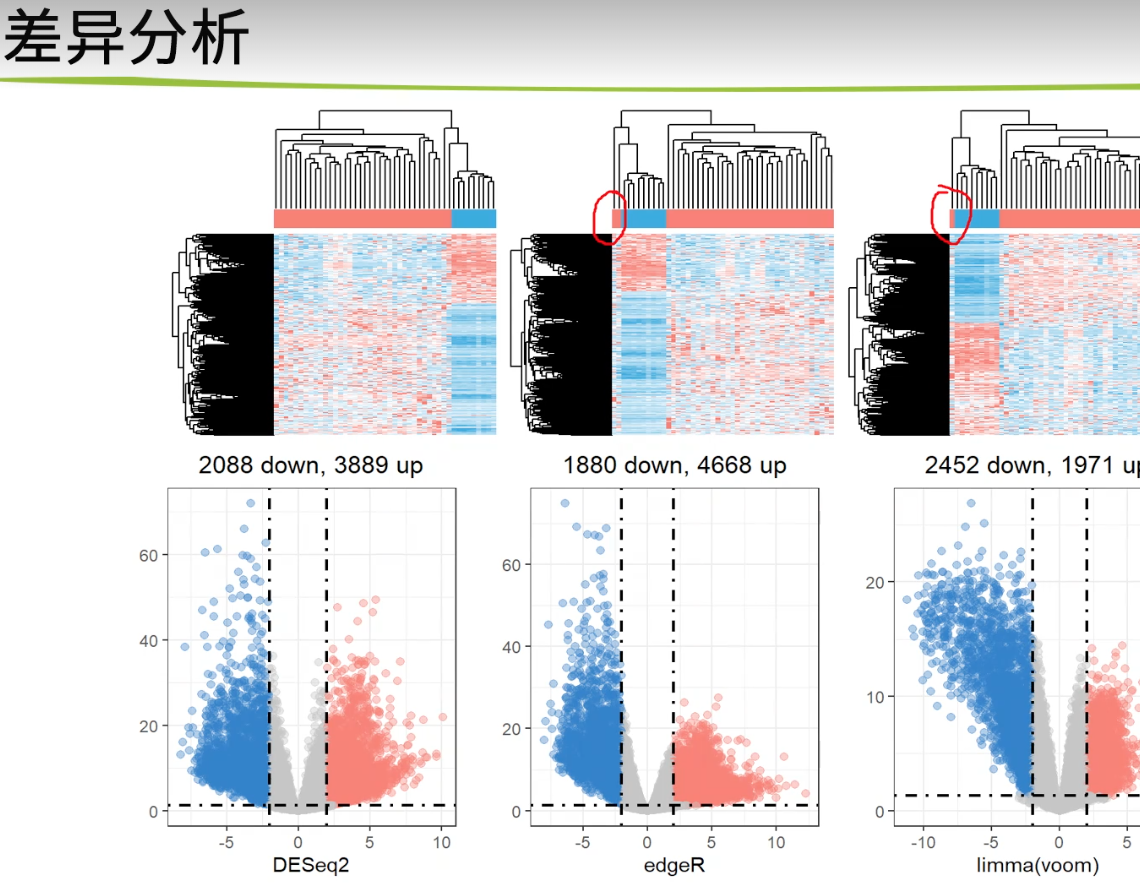
差异分析
counts矩阵必定是整数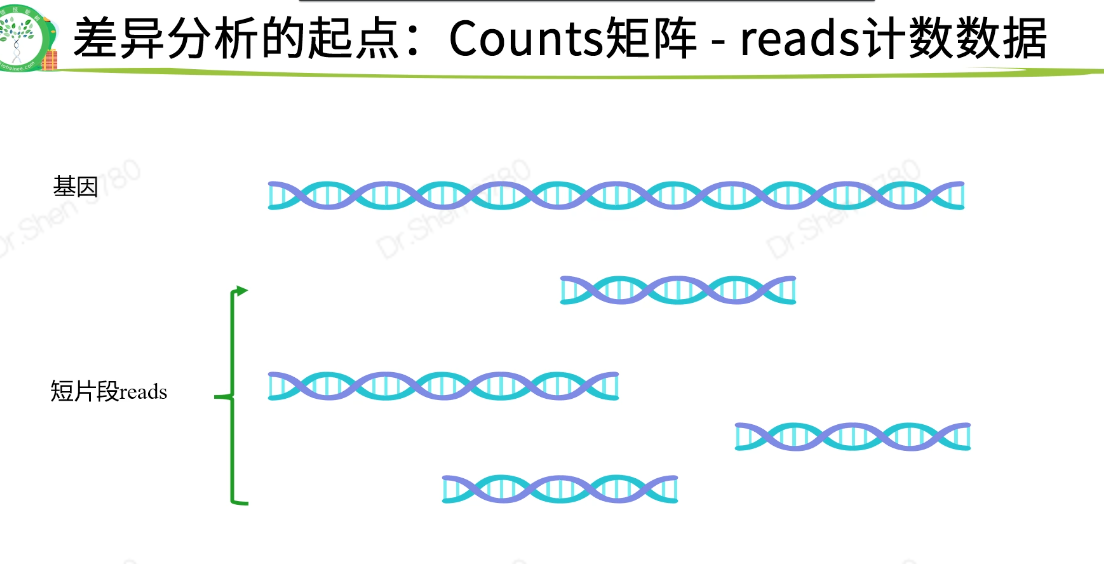
优选count数据,不得已才用以下的数据
在线数据库
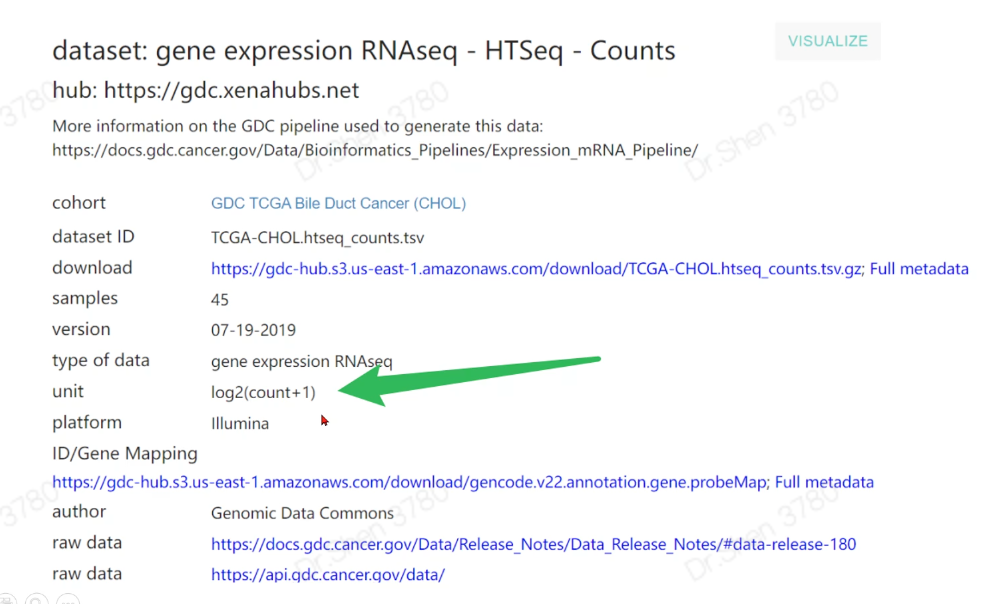
xena
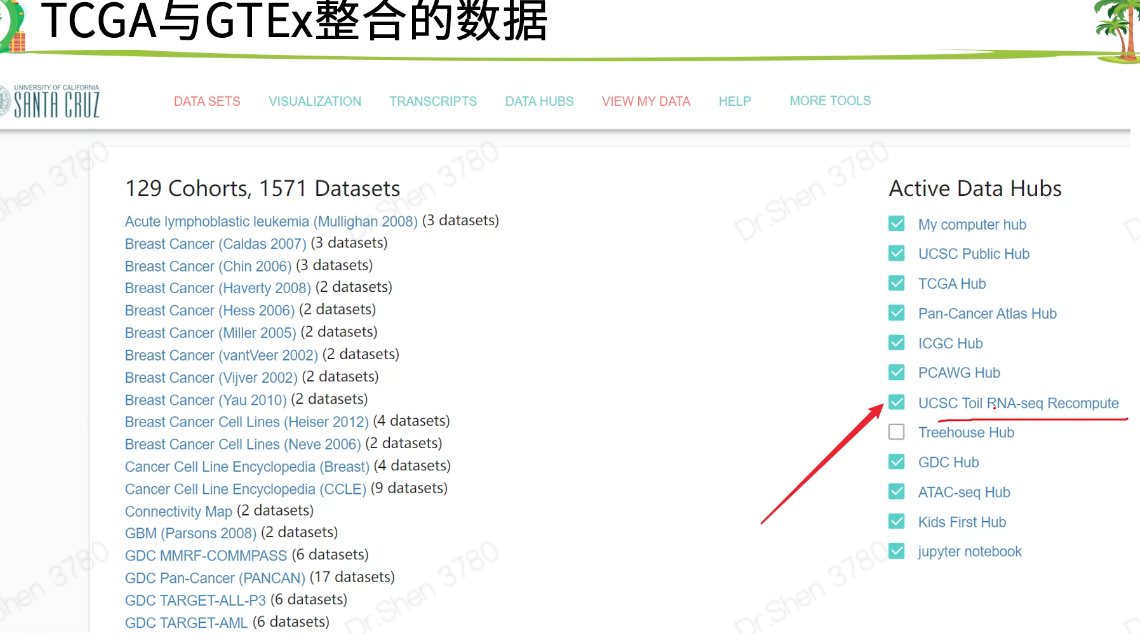
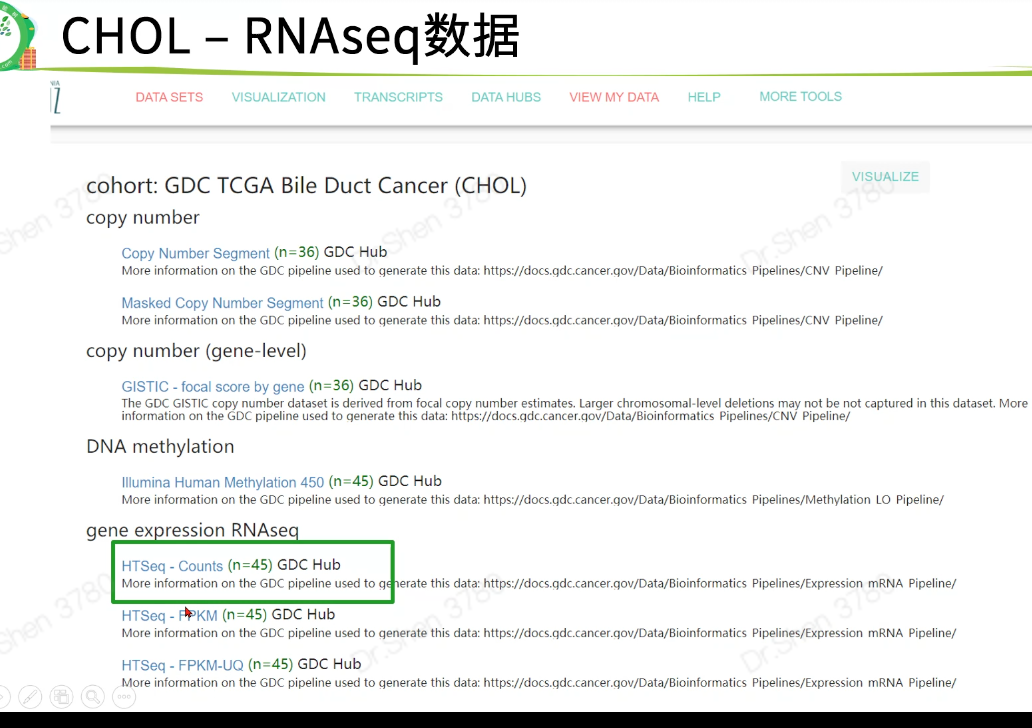
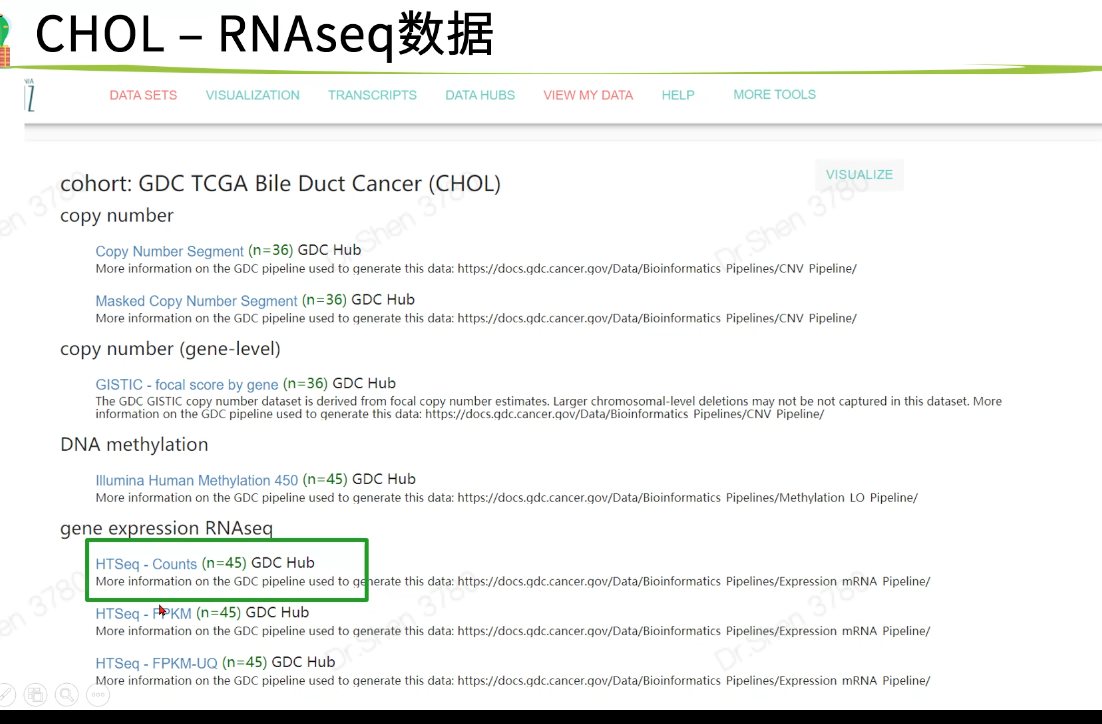
数据下载
数据下载

xena
proj = "TCGA-CHOL"if(F){download.file(url = paste0("https://gdc.xenahubs.net/download/",proj, ".htseq_counts.tsv.gz"),destfile = paste0(proj,".htseq_counts.tsv.gz"))download.file(url = paste0("https://gdc.xenahubs.net/download/",proj, ".GDC_phenotype.tsv.gz"),destfile = paste0(proj,".GDC_phenotype.tsv.gz"))download.file(url = paste0("https://gdc.xenahubs.net/download/",proj, ".survival.tsv"),destfile = paste0(proj,".survival.tsv"))}
读取和整理数据
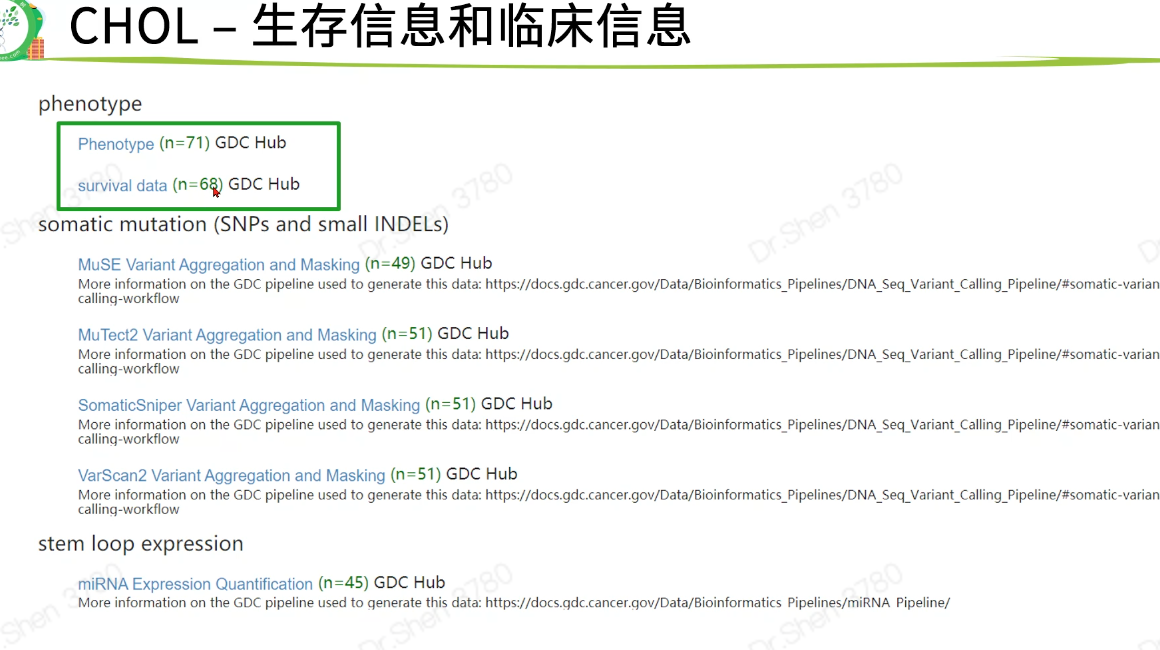
查看生存信息与临床信息
生存信息与临床信息在后面的差异分析里用不到,在此仅查看一下,到生存信息部分再整理
clinical = read.delim(paste0(proj,".GDC_phenotype.tsv.gz"),fill = T,header = T,sep = "\t")surv = read.delim(paste0(proj,".survival.tsv"),header = T)clinical[1:4,1:4]#> submitter_id.samples age_at_initial_pathologic_diagnosis#> 1 TCGA-ZH-A8Y2-01A 59#> 2 TCGA-ZH-A8Y7-01A 59#> 3 TCGA-W7-A93O-01A NA#> 4 TCGA-W7-A93O-11A NA#> albumin_result_lower_limit albumin_result_specified_value#> 1 NA NA#> 2 3.5 2.4#> 3 NA NA#> 4 NA NAhead(surv)#> sample OS X_PATIENT OS.time#> 1 TCGA-W5-AA31-01A 0 TCGA-W5-AA31 10#> 2 TCGA-W5-AA31-11A 0 TCGA-W5-AA31 10#> 3 TCGA-WD-A7RX-01A 1 TCGA-WD-A7RX 21#> 4 TCGA-YR-A95A-01A 1 TCGA-YR-A95A 26#> 5 TCGA-4G-AAZR-11A 1 TCGA-4G-AAZR 28#> 6 TCGA-4G-AAZR-01A 1 TCGA-4G-AAZR 28
查看矩阵
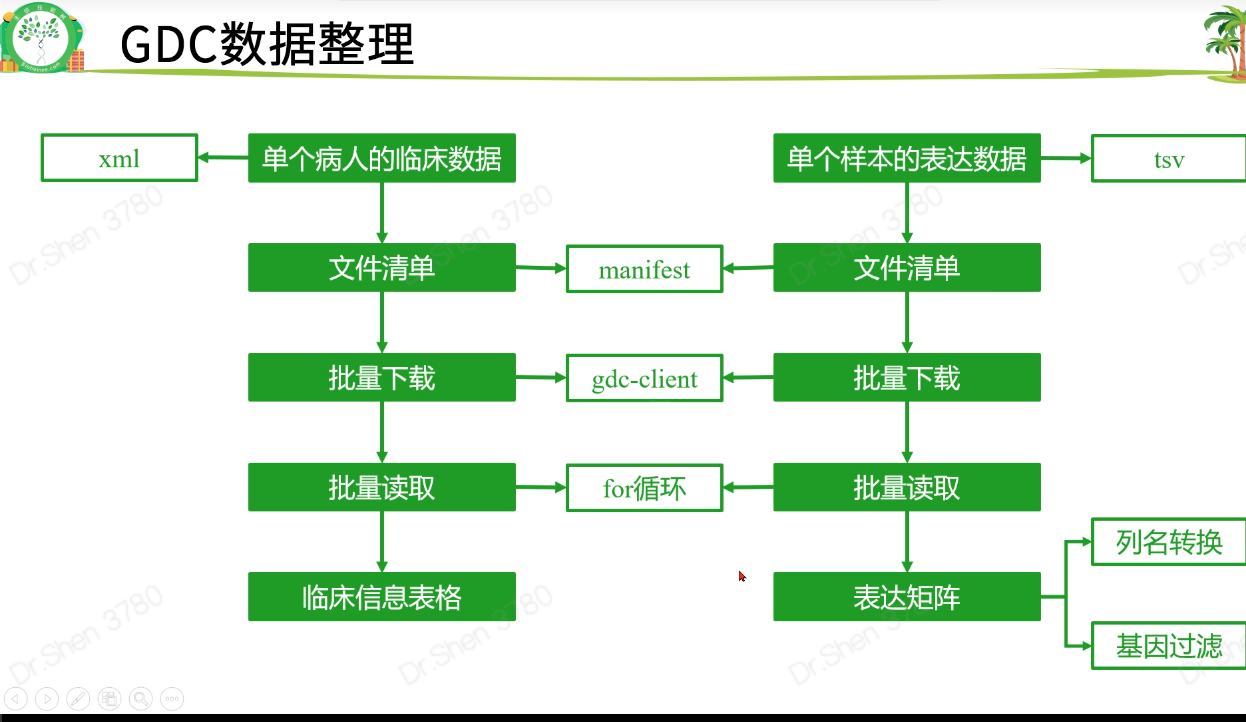
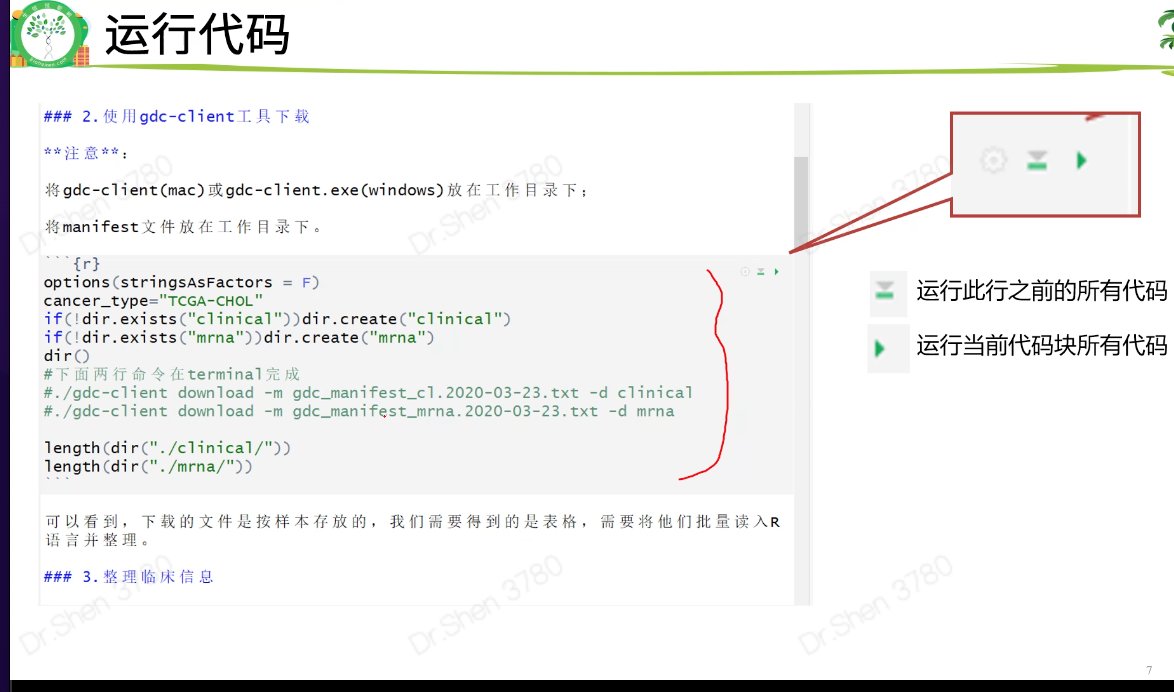
gdc下载的数据从此处开始衔接
dat = read.table(paste0(proj,".htseq_counts.tsv.gz"),check.names = F,row.names = 1,header = T)#逆转logdat = as.matrix(2^dat - 1)dat[1:4,1:4]#> TCGA-ZD-A8I3-01A TCGA-W5-AA2U-11A TCGA-W5-AA30-01A#> ENSG00000000003.13 5254 2476 5132#> ENSG00000000005.5 1 1 0#> ENSG00000000419.11 1212 655 1644#> ENSG00000000457.12 753 346 2652#> TCGA-W5-AA38-01A#> ENSG00000000003.13 8249#> ENSG00000000005.5 1#> ENSG00000000419.11 1696#> ENSG00000000457.12 519# 深坑一个dat[97,9]#> [1] 876as.character(dat[97,9]) #眼见不一定为实吧。#> [1] "875.999999999999"# 用apply转换为整数矩阵exp = apply(dat, 2, as.integer)exp[1:4,1:4] #行名消失#> TCGA-ZD-A8I3-01A TCGA-W5-AA2U-11A TCGA-W5-AA30-01A TCGA-W5-AA38-01A#> [1,] 5254 2476 5132 8249#> [2,] 1 1 0 1#> [3,] 1211 655 1643 1695#> [4,] 752 345 2652 519rownames(exp) = rownames(dat)exp[1:4,1:4]#> TCGA-ZD-A8I3-01A TCGA-W5-AA2U-11A TCGA-W5-AA30-01A#> ENSG00000000003.13 5254 2476 5132#> ENSG00000000005.5 1 1 0#> ENSG00000000419.11 1211 655 1643#> ENSG00000000457.12 752 345 2652#> TCGA-W5-AA38-01A#> ENSG00000000003.13 8249#> ENSG00000000005.5 1#> ENSG00000000419.11 1695#> ENSG00000000457.12 5193.表达矩阵行名ID转换library(stringr)head(rownames(exp))#> [1] "ENSG00000000003.13" "ENSG00000000005.5" "ENSG00000000419.11"#> [4] "ENSG00000000457.12" "ENSG00000000460.15" "ENSG00000000938.11"library(AnnoProbe)annoGene(rownames(exp),ID_type = "ENSEMBL") # 转换不了#> Warning in annoGene(rownames(exp), ID_type = "ENSEMBL"): 100% of input IDs are#> fail to annotate...#> [1] SYMBOL biotypes ENSEMBL chr start end#> <0 rows> (or 0-length row.names)rownames(exp) = str_split(rownames(exp),"\\.",simplify = T)[,1];head(rownames(exp))#> [1] "ENSG00000000003" "ENSG00000000005" "ENSG00000000419" "ENSG00000000457"#> [5] "ENSG00000000460" "ENSG00000000938"re = annoGene(rownames(exp),ID_type = "ENSEMBL");head(re)#> Warning in annoGene(rownames(exp), ID_type = "ENSEMBL"): 6.54% of input IDs are#> fail to annotate...#> SYMBOL biotypes ENSEMBL chr start#> 1 DDX11L1 transcribed_unprocessed_pseudogene ENSG00000223972 chr1 11869#> 2 WASH7P unprocessed_pseudogene ENSG00000227232 chr1 14404#> 3 MIR6859-1 miRNA ENSG00000278267 chr1 17369#> 4 MIR1302-2HG lncRNA ENSG00000243485 chr1 29554#> 6 FAM138A lncRNA ENSG00000237613 chr1 34554#> 7 OR4G4P unprocessed_pseudogene ENSG00000268020 chr1 52473#> end#> 1 14409#> 2 29570#> 3 17436#> 4 31109#> 6 36081#> 7 53312library(tinyarray)exp = trans_array(exp,ids = re,from = "ENSEMBL",to = "SYMBOL")exp[1:4,1:4]#> TCGA-ZD-A8I3-01A TCGA-W5-AA2U-11A TCGA-W5-AA30-01A TCGA-W5-AA38-01A#> DDX11L1 0 0 0 1#> WASH7P 81 10 146 54#> MIR6859-1 1 0 11 1#> MIR1302-2HG 0 0 0 0
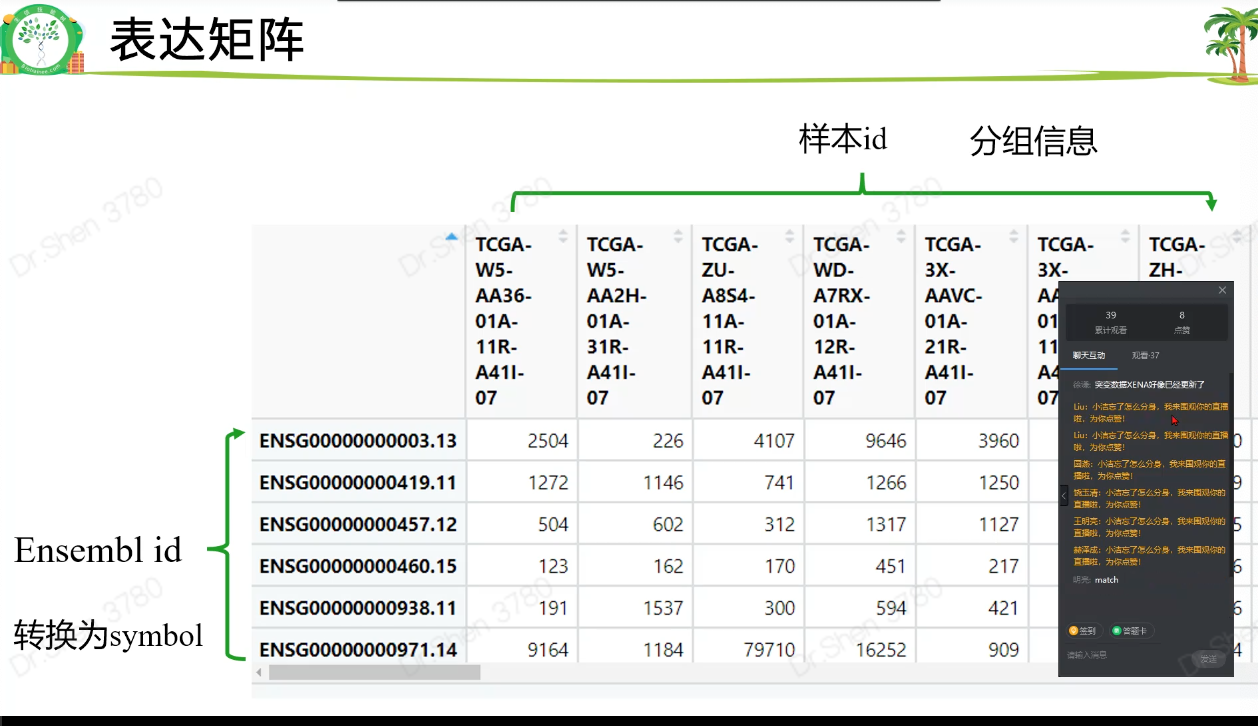

基因过滤
nrow(exp)#过滤之前基因数#> [1] 56514#常用过滤标准1:仅去除在所有样本里表达量都为零的基因exp1 = exp[rowSums(exp)>0,]nrow(exp1)#> [1] 48057#常用过滤标准2(推荐):仅保留在一半以上样本里表达的基因exp = exp[apply(exp, 1, function(x) sum(x > 0) >0.5*ncol(exp)), ]nrow(exp)#> [1] 28434
分组信息获取
table(str_sub(colnames(exp),14,15))#>#> 01 11#> 36 9Group = ifelse(as.numeric(str_sub(colnames(exp),14,15)) < 10,'tumor','normal')Group = factor(Group,levels = c("normal","tumor"))table(Group)#> Group#> normal tumor#> 9 36
保存数据
save(exp,Group,proj,clinical,surv,file = paste0(proj,".Rdata"))
回顾代码

x = round(rnorm(10),2)xsort(x,decreasing = T)#生成一个与x内容相同顺序不同的向量y = sample(x,10)xmatch(x,y) #谁的下标x[match(x,y)] #错y[match(x,y)]==xidentical(y[match(x,y)],x) # 两个数据是否一致names(x)=letters[1:10]class(x)library(stringr)x1 = str_split(sentences[2]," ")[[1]]ifelse(str_detect(x1,"th"),"hy","no")library(tibble)df1 = as.data.frame(x) %>%rownames_to_column(var = "letters")library(dplyr)arrange(df1,desc(x))df2 = data.frame(letters = letters,y = rnorm(26))df2merge(df1,df2,by = "letters")a = sample(letters[1:5],7,replace = T)aunique(a)a[!duplicated(a)]==unique(a)identical(a[!duplicated(a)],unique(a))x[1]x[x>1]x["a"]df1$y = iris$Sepal.Length[1:10]df1 = mutate(df1,z = iris$Sepal.Width[1:10])b = read.delim("TCGA-CHOL.GDC_phenotype.tsv.gz")load("gtf_gene.Rdata")save(b,file = "b.Rdata")pdf("a.pdf")pheatmap::pheatmap(volcano)dev.off()library(eoffice)f = pheatmap::pheatmap(volcano)topptx(f,"f.pptx")install.packages()BiocManager::install()devtools::install_github()library()


实现仅保留在一半以上样本里表达的基因的过滤标准2(推荐)
exp = exp[apply(exp, 1, function(x) sum(x > 0) > 0.5*ncol(exp)), ] #矩阵按行取子集,表达矩阵筛选基因nrow(exp)#对上述代码的理解,如下:#sum(逻辑值)代表T的个数,T是表达量大于0#sum(x>0)是某基因在多少个样本里有表达sum(x1 > 0) > 22.5sum(x2 > 0) > 22.5jianan = function(x) sum(x > 0) > 0.5*ncol(exp)x1 = as.numeric(exp[1,])x2 = as.numeric(exp[2,])jianan(x1)jianan(x2)#对矩阵的每一行(基因)做了一个函数的操作k = apply(exp, 1, jianan)#矩阵按行取子集,表达矩阵筛选基因k= apply(exp, 1, function(x) sum(x > 0) > 0.5*ncol(exp));table(k)exp = exp[k,]dim(exp)exp = exp[apply(exp, 1, function(x) sum(x > 0) > 0.5*ncol(exp)), ]
gdcRNAtools
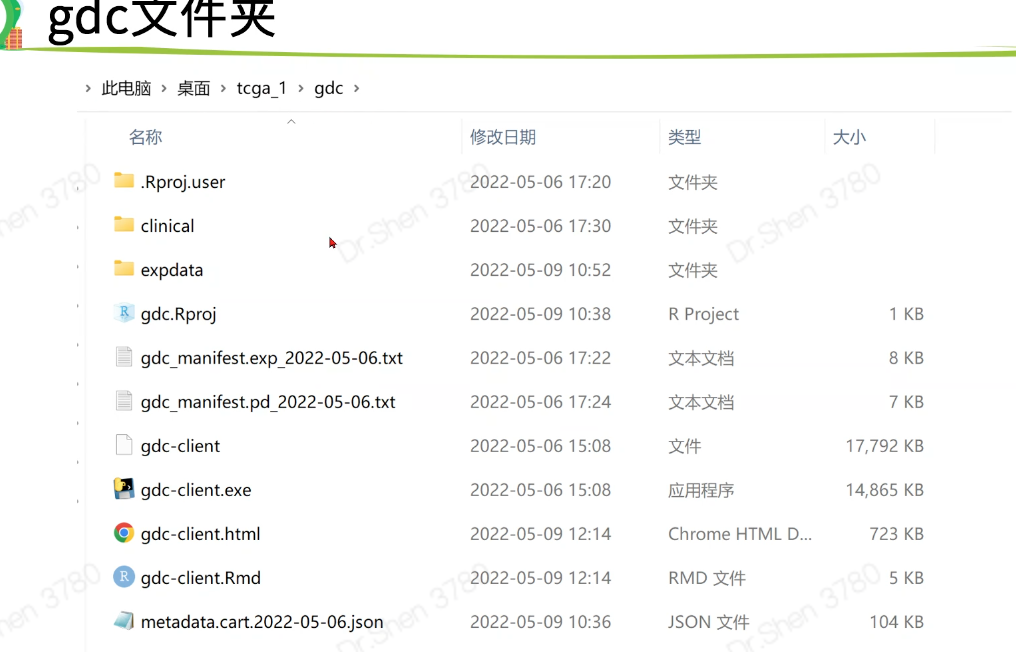
gdc-client

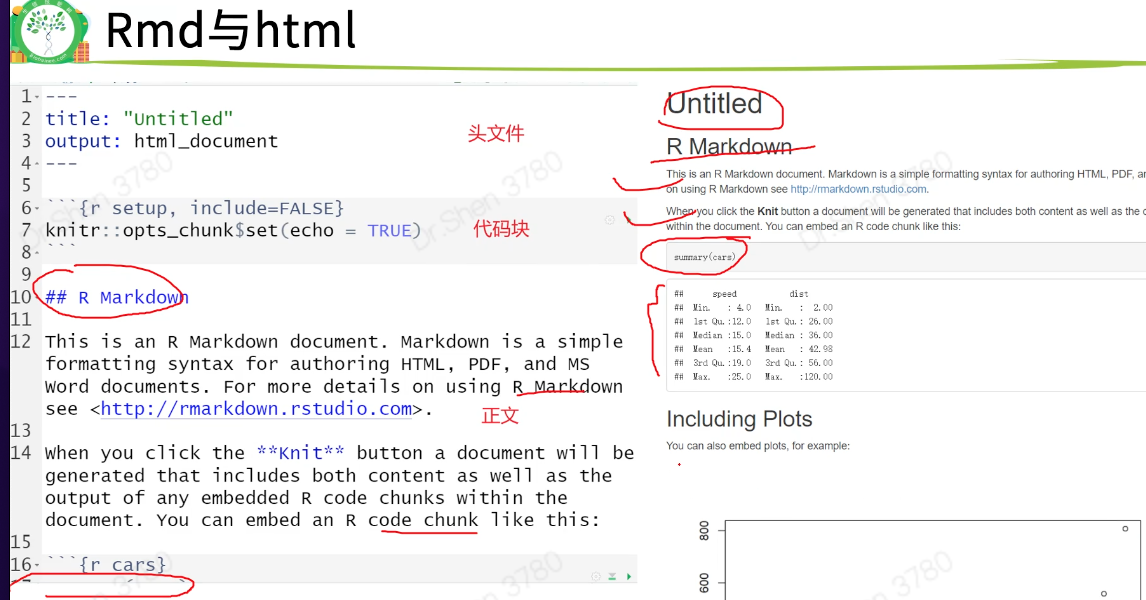
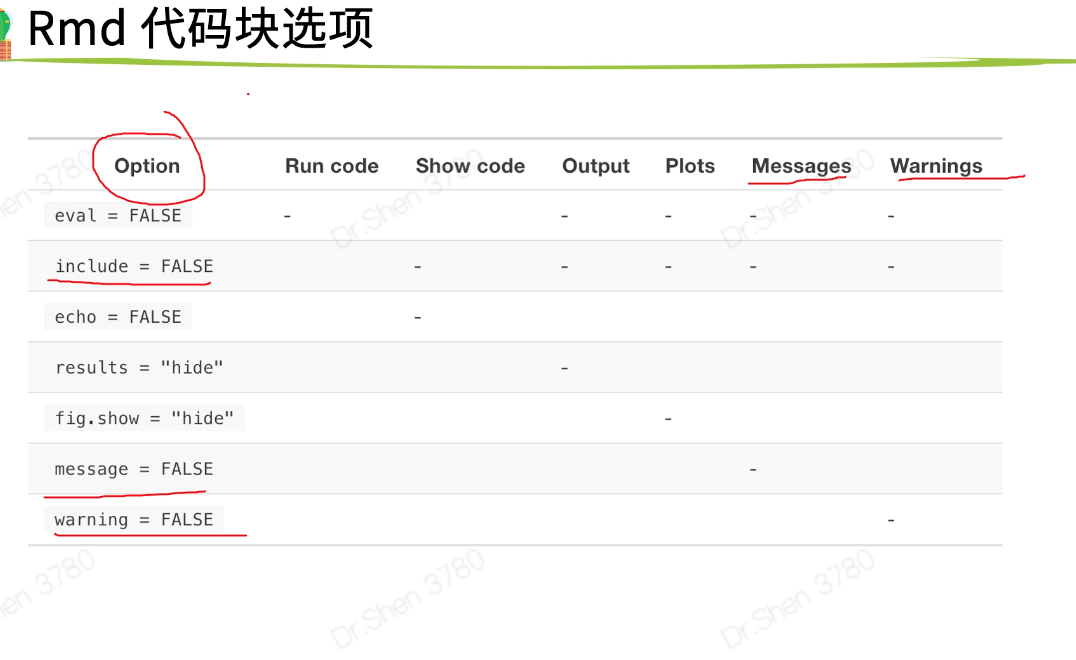
R markdown

若有收获,就点个赞吧
0 人点赞
