1. Introduction of SPAdes:
1.1 Purpose & Function:
SPAdes is used primarily for assembling bacterial genomes for single-celled sequencing, but also non-single-celled sequencing data.
The input data can be Illumina, IonTorrent reads, PacBio, Sanger Reads, or some contigs sequences can be input as long reads.
This software can receive multiple sets of paired-end, mate- match, and Unpaired reads simultaneously. At the same time, the software has a separate module for the assembly of heterozygous genomes
See the introduction to see: https://www.jianshu.com/p/08d62d9d1a1f
1.2 Installation
wgethttp://cab.spbu.ru/files/release3.13.0/SPAdes-3.13.0-Linux.tar.gztar - XZF SPAdes - 3.13.0 - Linux. Tar. Gzcd SPAdes - 3.13.0 - Linux/bin /
Successful test installation:
Python./bin /spades.py --test
(Note that you need to call Python before you start spades.py.)
1.3 Basic parameters:
-o Specify the output directory. Required options. —isolate This flag is highly recommended for high-coverage isolate and multi-cell data —sc This flag is required for (single-cell) data —meta This flag is required for metagenomic sample data —plasmid -1 File with forwarding reading. -2 File with reverse read. -s Files that are not read in pairs. (Unpaired reads generated by subsample-S parameter) — file_name> Contains the file that is merged into the paired read. -t (or —threads )Threads. The default value is 16 -k A comma-separated list of k-mer sizes to use (all values must be odd, less than 128, and listed in ascending order). If —sc is set, the default value is 21,33,55. For multicellular data sets, the K value is automatically selected using the maximum read length (for more information, see the notes for assembly length Illumina paired readings) —cov-cutoff To read coverage cutoff value. Must be a positive floating-point value, or “auto” or “off.” The default value is “off”. When set to “auto,” SPAdes automatically calculates the coverage threshold using the conservative policy. Note that metaSPAdes does not support this option.
Note:
-s: # with files that are not read in pairs. (Unpaired reads generated by subsample-S parameter)
-k: int, int,… K-mer sizes separated by commas. These values must be odd, less than 128, and in ascending order. If the —sc parameter is used, the default value is 21,33,55. If there is no — SC parameter, the program automatically selects the K-mer parameter based on the read length.
— meta:
This flag is recommended for metagenomic data sets. Currently, metaSPAdes only supports reads that match the ends of the short-reading library.
— sc: For single-cell data.
— Service Organizations: They are good for capturing data from single or multiple cells with high coverage.
2. Process and results
python spades.py -1 /public/home/yuanmy/q89/out.R1.fq.gz -2 /public/home/yuanmy/q89/out.R2.fq.gz -o ./out2/
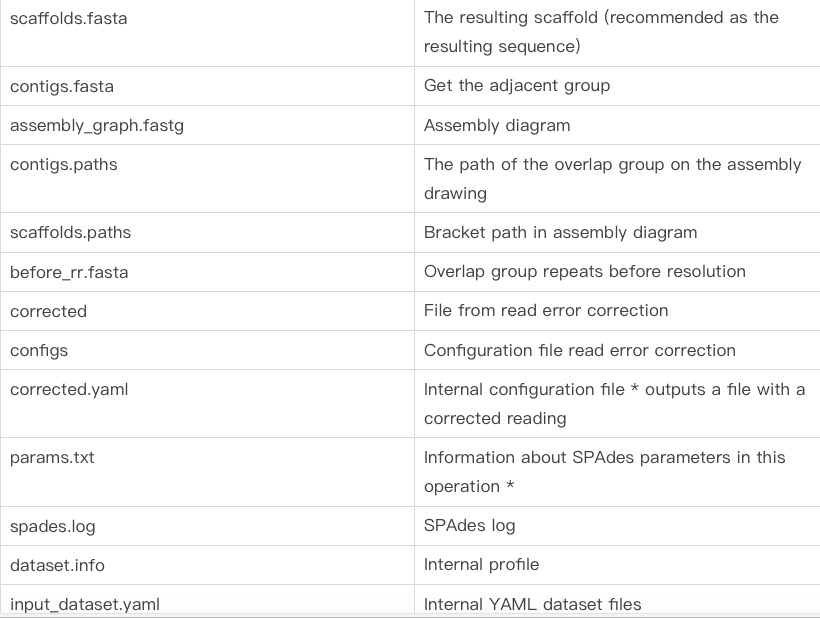
The contents of the results folder:
Analysis of results: 1. No significant difference was found between contigs and scaffold files. 2.K value selection: KmerGenie can perform K-MER analysis and genome size assessment. The greatest advantage of KmerGenie is that it can realize automatic analysis under multiple preset K-mers. In addition to regular k-mer frequency statistics, it can also automatically calculate genome size based on different K-mers, and evaluate an optimal assembly K-mer value as an alternative for genome assembly. After Kmergene evaluates the optimal assembly K value, it is assembled (which can be optimized later).

若有收获,就点个赞吧
0 人点赞
