1. Download and install
wget http: / /https://github.com/broadinstitute/pilon/releases/download/v1.23/pilon-1.23.jarJava - Xmx10G - jar pilon - 1.23. The jarJava - Xmx10G - jar pilon - 1.22. The jar
2. Prepare the data
1. Genome files Scaffold. fa assembled with three generations of data2. Bam. sort file built with the assembled fasta as an index. Can be divided into 4 KINDS of BAM files
| bam | 4 kinds of bam files |
|---|---|
| The bam. sort file of the subsample handles the resulting double-ended FQ | subfq1,subfq2>alignsort1.bam |
| SPAdes are assembled with corrected reads | meta-cor1.bam |
| SPAdes assembled unpaired reads | unpaired1.bam |
| Mardup labeled BAM file after PCR repetition | cor-markdup1.bam |
3. Comparison
The index
Bwa index -p (prefix) meta1/meta1-cor meta1. Fasta /meta1-cor. Fasta
The comparison sort
bwa mem -t 16 index subv1-1.fg subv1-2.fq |samtools sort -@ 10 -O bam -o alignsort1.bambwa mem -t 16 index meta1-cor1.fq meta1-cor2.fq |samtools sort -@ 10 -O bam -o meta-cor1.bam
Compare and index BAM files
samtools index -@ 10 alignsort1.bam
sambamba markup -t 10 align.bam align_markup.bam
Important parameters needed to improve genomic assembly :(select —fix, not — VCF, note —unpaired parameter)
**
Java-xmx {$x} g-jar pilon-1.23.jar --genome meta1.fasta --frags align_markdup1.bam --fix SNPS,indels --output pilonpolished1 --outdir pilon &> pilon2.log
The important paramete**rs **usage
-Xmx{$x}GDetermine the memory size based on the genome size--bamThe type of BAM input can be automatically identified--fragsEnter SubfQ or SPAdes corrected fq reads--duplicatesThe BAM file for Markdup--unpairedEnter Unpaired reads in the SPAdes corrected file--fixChoose to repair the non-strategy (single base error) SNPs (small Indel) Indels, fasta output of the ambiguous base (AMB), and allow local recombination to open up new white breaks
4. Experimental design
Strategy 1:
Subfq reads constructed index alignment to obtain the BAM file with the meta-FastA file obtained after the first SPAdes assembly.
Strategy 2:
Corrected reads are compared with the BAM obtained with the FASTa file obtained after the second Spades.
Strategy 3:
— FRAGS bAM file marker PCR repeat markup. bAM file, — Unpaired Bam generated by Unpaired Unpaired reads.
Strategy 4:
— Frags Strategy 3 markup. bam file, no input of Unpaired Bam file
Strategy 5:
— FRAGS BAM file in strategy 2, — Unpaired Bam generated by Unpaired unpaired reads.
Quast evaluates these several Polish results as indicators
5. Interpretation of results:
Fasta file after different Polish strategies
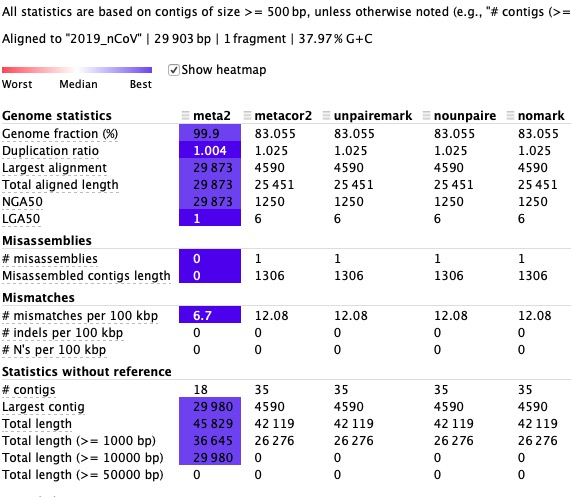
The comparison can be seen as follows:
1.If SPAdes are re-assembled (only corrected reads are re-assembled), genomic coverage decreases after assembly and increases per 100kbp misalignment.
2.This decrease cannot be further improved by increasing the coverage of unpaired BAM files generated by SPAdes and corrected reads at the first pass (compared with fasta generated by unpaired reads at the first pass).
Conclusion: SPAdes does better with a single assembly than with corrected reads.
Question: Can the unpaired BAM from unpaired reads assembled in the first time improve the fastA file formed in the first time?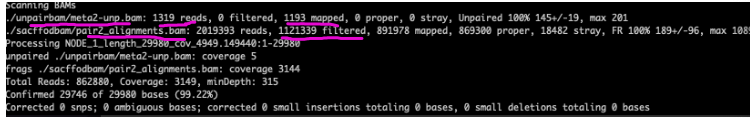
The figure is the report of the Pilon operation. It can be seen that there are 1193reads in the pilon process of the Unpaired Bam file, so it is assumed that it is useful and further exploration will be carried out.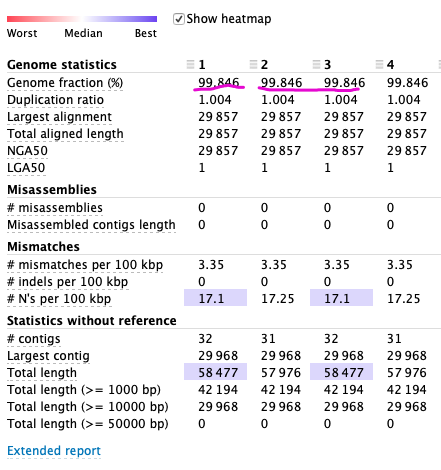
Comparison: From left to right are 1. Not polish2. Polish only enter —frags 3. Polish enter —frags and — unpaired, 4. Is corrected. Bam and unpaired. Bam
Conclusion: The fasta assembled best without the second SPAdes.The effect of SPAdes (corrected reads only) and Polish of Unpaired reads were better than that of the second time.

若有收获,就点个赞吧
0 人点赞
