1. What is the Fsatq format?
Is a text-based file format that stores the quality of biological sequences and corresponding bases (or amino acids). Originally developed by the Wellcome Trust Sanger Institute, it has become the de-facto standard for storing high-throughput sequencing data
2.General description of Fastq format
Each sequence is represented by 4 lines of characters, for example, the first pair of data in this analysis (in the format of Illumina results)
@A00821:275:HWMMWDSXX:4:1101:1353:27821 1:N:0:ATTACTCG+TAGATCGC AGTGCTGCTAAAAAGAATAACTTACCTTTTAAGTTGACATGTGCAACTACTAGACAAGTTGTTAATGTTGTAACAACAAAGATAGCACTTAAGGGTGGTAGATCGGAAGAGCACCACGTCTGAACTCCAGTCACATTACTCGATCTCGTA + FFFFFFFFF:FFFFFFFFFFF:FFF:F:FFF
The first line:
You must start with “@”, followed by a unique sequence ID identifier, followed by an optional sequence description, separated by Spaces.
The second line:
Sequence character (nucleic acid is [AGCTN]+).
The third line:
You must start with a “+” followed by an optional ID identifier and optional description. If there is anything after the “+”, it must be the same as the first line after the “@”.
The fourth line:
Base mass character, each character corresponds to the base or amino acid mass of the corresponding position in the second line, which can be converted into a base mass score according to certain rules, and the base mass score can reflect the error rate of the base. This line must have the same number of characters as the second line. The specific relationship between characters and the error rate is described below.
3.The first line of Fastq is as follows:
@A00821:275:HWMMWDSXX:4:1101:1353:27821 1:N:0:ATTACTCG+TAGATCGC
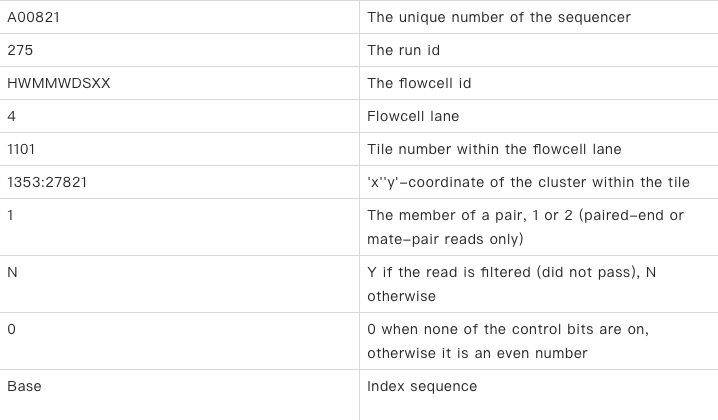
4. The calculation of sequencing mass fraction in the fourth line of Fastq
Phred calculates several parameters related to the size and resolution of the crest, and based on these parameters, the base mass score is extracted from a large query table.
Different sequencing reagents and machines use different query tables. To save disk space, quality scores (which can take up to two characters) are converted to a single character representation according to certain rules (Phred+33 or Phred+64).
Different software uses different schemes for the quality coding of each base, and there are currently 5 schemes:
The relationship between base error rate and mass score is as follows:
Qphred = -10log10p Qillumina-prior to V. 1.4 = -10log10(p/(1-p))

若有收获,就点个赞吧
0 人点赞
