
1. Samtools introduction:
1.1: Purpose function:
Purpose:
Use SAMtools to convert SAM files to BAM files, sort, and index them
See: https://blog.csdn.net/xiaomotong123/article/details/106504192/
Function
1.2: Installation:
conda install -y samtools
2.sam to bam
Samtools View-@8-s 2d.sam-o 2d.bam (single Sam file)(Batch processing)INDEX=./bwa_index/2019-nCoV.fastafor i in $(ls *.sam)doi=${i/.sam/}samtools view -@ 8 -S $i.sam -1b -o $i.bamdone
3.Samtools sort
Samtools sort-@8-L 5-o 2d.bam. Sort 2d.bam (single BAM)samtools sort -@ 8 -l 5 -o {$i}.bam.sort {$i}.bamsamtools sort -@ 8 -l 5 -n -o P10.bam.sort XGRNAMIX1-10-N-RNA_L3.bam-@ Number of threads- L compression level- O output form-n sorted by read Name
The BAM file sort gets smaller:
BAM file is a compressed binary file, after sorting the contents of the file, similar contents are arranged together, so the compression ratio of the file is improved, so the BAM file after sorting becomes smaller, and the corresponding SAM file is a plain text file.
Sorting the SAM file will not change the file size. Reads with no alignment are placed at the end of the BAM file after samTools sorts it.
4.Samtools index
samtools index -@ 8 2nd.bam.sort 2nd.bam.indexsamtools index -@ 8 {$i}.bam.sort {$i}.bam.index
5.Samtools flagstat
samtools flagstat -@ 8 2nd.bam.sortsamtools flagstat -@ 8 {$i}.bam.sort
6. Compose scripts
#! /bin/bash# Concatenates the view, sort, and index commands of SamToolsFor I in stores file namesdosamtools view -@ 8 -S /$i.sam -1b -o /$i.bamsamtools sort -@ 8 -l 5 -o /$i.bam.sort /aligned/$i.bamsamtools index -@ 8 /$i.bam.sort /$i.bam.indexdone
7. Interpretation of SAM-BAM-sort-index-Flagstat results
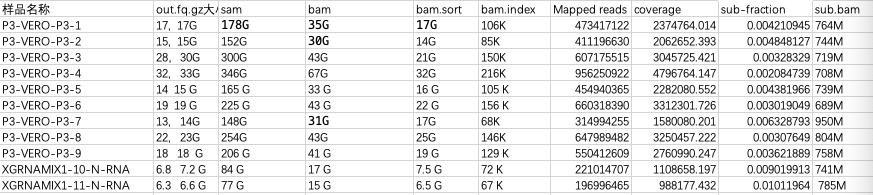
7.1: Sam, BAM, bam. Sort file size
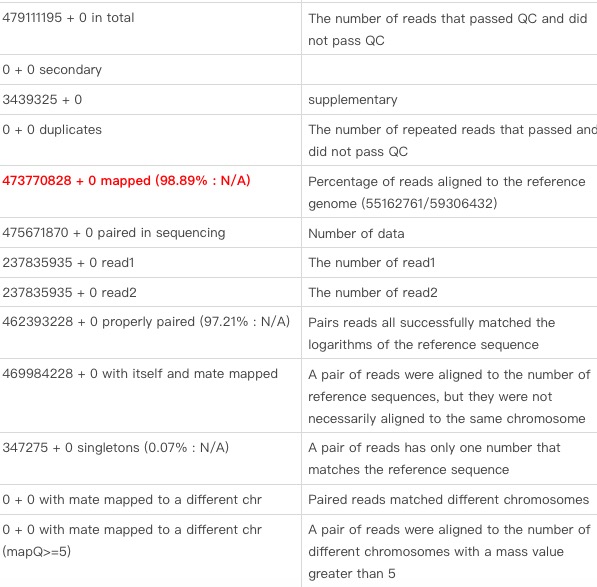
7.2: Flagstata results (note the results in red)

若有收获,就点个赞吧
0 人点赞
