Result 3: Package the pipeline :
(1)Goals:
Input the original paired-end fastq files、index files、subsampling fraction and assembling strategies, the output file will be the finally assembled fasta files with only single contigs and the whole process taking around 7 hours for each sample.
Usage:
Run the following shell from the command line:#!/bin/bash#SBATCH -e subfqall.err#SBATCH -o subfqall.out#SBATCH -J subfqall#SBATCH --ntasks-per-node=2#SBATCH -N 1#SBATCH -p sugon#SBATCH -t 120:00:00
python ``ymy.py`` ``--filea`` ./P3-VERO-P3-1-vero_L4_1.fq.gz ``--fileb`` ./P3-VERO-P3-1-vero_L4_2.fq.gz ``--index`` ./2019-nCoV.fasta ``--fraction`` 0.0036 ``--startegy`` meta
(2)—help**: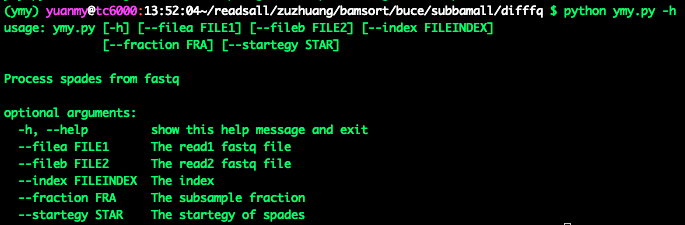
(3)Code:**
#!/usr/bin/python#-*- coding:utf-8 -*-import argparseimport osdef _argparse():parser = argparse.ArgumentParser(description='Process spades from fastq')parser.add_argument('--filea', dest='file1', default='./P3-VERO-P3-1-vero_L4_1.fq.gz', help="The read1 fastq file")parser.add_argument('--fileb', dest='file2', default='./P3-VERO-P3-1-vero_L4_2.fq.gz', help="The read2 fastq file")parser.add_argument('--index', dest='fileindex', default='./2019-nCoV.fasta', help="The index")parser.add_argument('--fraction', dest='fra', default='0.00036', help="The subsample fraction")parser.add_argument('--startegy', dest='star', default='--meta', help="The startegy of spades")return parser.parse_args()def zuzhuang(x,y,z,w,t):'''os.system(f"fastp -i {x} -I {y} -o out1.fastq.gz -O out2.fastq.gz")os.system(f"bwa mem -t 20 {z} out1.fq.gz out2.fq.gz | samtools sort -@ 10 -O bam -o sorted.bam")os.system(f"samtools index -@ 8 sorted.bam sorted.bam.bai")os.system(f"sambamba view -t 10 -h -s {w} sorted.bam -o subsorted.bam")os.system(f"samtools fastq -s -f 3 -1 paired1.fq -2 paired2.fq subsorted.bam")os.system(f"spades.py -{t} -1 paired1.fq -2 paired2.fq -o outputfile")'''os.system("fastp -i {x} -I {y} -o out1.fastq.gz -O out2.fastq.gz".format(x=x, y=y))os.system("bwa mem -t 20 {z} out1.fq.gz out2.fq.gz | samtools sort -@ 10 -O bam -o sorted.bam".format(z = z))os.system("samtools index -@ 8 sorted.bam sorted.bam.bai")os.system("sambamba view -t 10 -h -s {w} sorted.bam -o subsorted.bam".format(w = w))os.system("samtools fastq -s -f 3 -1 paired1.fq -2 paired2.fq subsorted.bam")os.system("spades.py --{t} -1 paired1.fq -2 paired2.fq -o outputfile".format(t = t))#return outputfileprint("all finished!")def main():parser = _argparse()zuzhuang(parser.file1, parser.file2, parser.fileindex, parser.fra, parser.star)if __name__ == '__main__':main()
(4)Performance on 11 samples :
Parameter selection:
Filtering parameter during subsampling : -f 3
Subsampling coverage selection: 1000-10000
SPAdes strategies: —meta,**
Assembled results of 11 samples:
Each sample only has one contig in the assemblie. Some indicators, as the coverage of reference genome, the miss-assembled rates and the error rate are more reliable than before.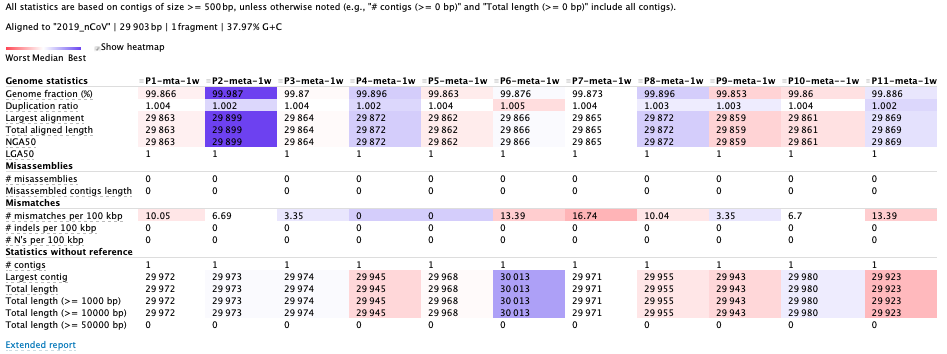

若有收获,就点个赞吧
0 人点赞
