
1.Software download and install:
git clone [https://github.com/BimberLab/DISCVRSeq/](https://github.com/BimberLab/DISCVRSeq/)java -jar DISCVRseq.jar VariantQC --help
2.Details of the most important parameters:
_-O _``File to which the report should be written_-R _``Reference sequence file_-V _``A VCF file containing variants
Note: The VCF file is obtained from the Pilon step. And in Pilon, we choose the function of identifying variation rather than the function of polishing the assembly result. The input of the BAM file is the subsample of the original BAM file generated by the FASTQ file, and the genome input is the reference genome instead of the assembled fasta file.
3.Pipeline:
3.1 Install Picard and index the reference sequence:
Before using Variant QC, some preparations are required.
First, index the reference sequence and variant VCF files respectively: Index the reference sequence, which is generally done before using GATK:
conda install -c bioconda picardsamtools faidx meta1.fastajava -jar picard.jar CreateSequenceDictionary R=meta1.fasta O=meta1.dict
3.2 Install GATK and index the variation file:
Then the index is performed for the variation file to generate xx. vcf. Idx file. GATK4.0 is used here:
conda install -y gatk<br />Wget -c [https://github.com/broadinstitute/gatk/releases/download/4.1.9.0/gatk-4.1.9.0.zipUnzip](https://github.com/broadinstitute/gatk/releases/download/4.1.9.0/gatk-4.1.9.0.zipUnzip) gatk - 4.1.9.0. Zip<br />cd gatk 4.1.9.0 /chmod 777 gatk./gatk --list
Build Index on the VCF file
Java-jar./ Gatk-4.1.9.0 / Gatk-package-4.1.9.0-local.jarIndexFeatureFile -i subfq1. vcf
3.3: VariantQC
Start using Variant QC when the two steps above are ready. The running time of Variant QC is related to the size and number of the variants of VCF file.
Java-jar discvrseq-1.18. jar variantQC-o van1. Html-r meta1. Fasta-- maxgs 500-v subfq1.vcf
4. Variant QC Output Report:
4.1 The html report has 4 aspects:
A complete example html report contains a series of tables or graphs summarizing four aspects of the data, namely Entire VCF, By Contig, By Sample, and By Filter Type.
4.2 Each aspect have 6 kinds of statistics:
As for each aspect, statistics of different types of VCF variation files are provided. Here I intercepted the statistics of the Entire VCF, and use the results of sample 1.
- Variant Summary: A table displaying a summary of the total variants by type (SNP, insertion, deletion, MNP etc).
- Variant Type: a bar plot summarizing variant by type (SNP, insertion, deletion, MNP etc).
- Genotype Summary: A table summarizing the total called/non-called genotypes.
- SNP/Indel Summary.
- Ti/Tv Data: A table displaying a summary of transition and transversion mutations in the dataset.
- Filter Type: A bar plot summarizing variants by filtering.
4.3 The result example of Sample 1
4.3.1 Variant Summary
Here, we can see various statistics that are called, filtered, and raw. There are up to 27 columns of statistics, such as total loci, called loci, the total number of variants, and so on (We only show 11 columns of them here). Columns can also be customized using the Configure Columns function, and then use the Plot function to Plot the columns you want.
The total number of variant loci is 3.
**
4.3.2 Variant Type
Here is the type of variation, which can be presented according to the number of reads corresponding to the variation or its corresponding percentage. So we can see the number of mutations in the range of SNPs, Insertions, Deletions. This image can also be output at any time using the Exportplot function.
It can be seen here that all the identified variations are SNPs with three loci.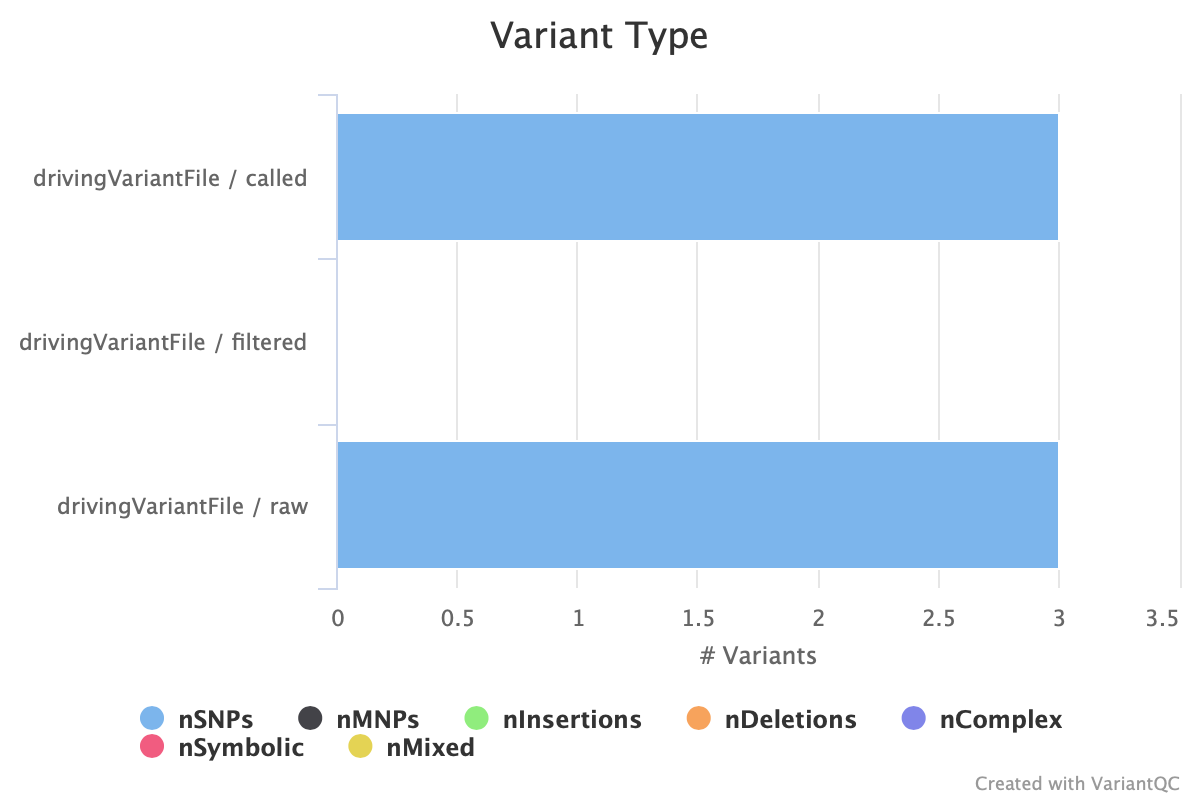
4.3.3 Genotype Summary
The following is the statistical summary of genotypes, showing the number of called and uncalled genotypes.

4.3.4 SNP/Indel Summary
The following is the statistical summary of SNPs and Indels, including information on the number of SNPs, singleton SNPs, Indels, and so on.
5. General information of variation:
In all of the VCF variation files of 11 samples, the number of SNPs in the 7th sample is at most 5, while the number of SNPs in the rest samples is less than 3. There is no other variation such as deletion indel.

若有收获,就点个赞吧
0 人点赞
