数据准备
首先还是同样的读入数据,进行一定的处理。我们同样用 VEP 注释后的 maf 文件,然后取出需要用到的几列
rm(list=ls())options(stringsAsFactors = F)library(dplyr)library(stringr)# 读入数据laml = read.maf('./7.annotation/vep/VEP_merge.maf')laml@data=laml@data[!grepl('^MT-',laml@data$Hugo_Symbol),]# 增加一列t_vaf,即肿瘤样本中突变位点的覆盖深度t_alt_count占测序覆盖深度t_depth的比值laml@data$t_vaf = (laml@data$t_alt_count/laml@data$t_depth)unique(laml@data$Tumor_Sample_Barcode)getSampleSummary(laml)getGeneSummary(laml)getFields(laml)mut = laml@data[laml@data$t_alt_count >= 5 &laml@data$t_vaf >= 0.05, c("Hugo_Symbol","Chromosome","Start_Position","Tumor_Sample_Barcode","t_vaf")]mut$patient = substr(mut$Tumor_Sample_Barcode, 1, 5)
最后的数据框如下:
> mut[1:6,1:6]Hugo_Symbol Chromosome Start_Position Tumor_Sample_Barcode t_vaf patient1: ADGRB2 chr1 31756671 case1_biorep_A_techrep 0.06976744 case12: PTPRF chr1 43569766 case1_biorep_A_techrep 0.15000000 case13: TCTEX1D1 chr1 66770463 case1_biorep_A_techrep 0.08816121 case14: C1orf68 chr1 152719922 case1_biorep_A_techrep 0.24267782 case15: ARHGEF11 chr1 156936822 case1_biorep_A_techrep 0.27027027 case16: OCLM chr1 186401109 case1_biorep_A_techrep 0.09800919 case1
数据处理
然后进行处理,得到每一个病人的 4 个样本(列)和突变基因(行)的矩阵,因为用 lapply 进行循环处理,所以最后 6 个病人的 6 个矩阵会组合成一个列表
pid = unique(mut$patient)all_snv = lapply(pid , function(p){# p='case1'print(p)mat=unique(mut[mut$patient %in% p,c("Tumor_Sample_Barcode",'Hugo_Symbol')])mat$tmp = 1# 长变扁mat = spread(mat,Tumor_Sample_Barcode,tmp,fill = 0)class(mat)mat = as.data.frame(mat)rownames(mat) = mat$Hugo_Symbolmat=mat[,-1]dat = mat[order(mat[,1],mat[,2],mat[,3],mat[,4]),]return(dat)})
如病人 case1 的矩阵如下(也就是上一节画热图的矩阵):
> datcase1_biorep_A_techrep case1_biorep_B case1_biorep_C case1_techrep_2ETHE1 0 0 0 1LRTM2 0 0 0 1CFHR1 0 0 1 0GLRA1 0 0 1 0SMAD4 0 0 1 0DCP1B 0 1 0 0FBXW12 0 1 0 0MORC2 0 1 0 0......
突变分类
trunk_gene = unlist(sapply(all_snv, function(x) rownames(x[rowSums(x) == 4,])))branch_gene = unlist(sapply(all_snv, function(x) rownames(x[rowSums(x) == 3|2,])))private_gene = unlist(sapply(all_snv, function(x) rownames(x[rowSums(x) == 1,])))
每个向量就记录了该类型突变的基因:
> trunk_gene[1] "A4GNT" "ARHGEF11" "ATRX" "ATXN1" "BUD31" "C1orf68" "CALM1"[8] "CARD14" "CCDC73" "COL6A3" "DENND1A" "DMD" "DNAH17" "DRG2"......[134] "ALMS1" "C2orf78" "CACNA1D" "FNDC3B" "MAK" "OR1N1" "PGC"[141] "PIK3CA" "SPHKAP" "SYNPO2" "TBRG4" "TTN" "ZNF571" "ZXDA"> branch_gene[1] "ETHE1" "LRTM2" "CFHR1" "GLRA1" "SMAD4" "DCP1B" "FBXW12"[8] "MORC2" "MPHOSPH9" "PCDHGA4" "POLG" "PRICKLE2" "UBA2" "MADCAM1"......[344] "ALMS1" "C2orf78" "CACNA1D" "FNDC3B" "MAK" "OR1N1" "PGC"[351] "PIK3CA" "SPHKAP" "SYNPO2" "TBRG4" "TTN" "ZNF571" "ZXDA"> private_gene[1] "ETHE1" "LRTM2" "CFHR1" "GLRA1" "SMAD4" "DCP1B" "FBXW12"[8] "MORC2" "MPHOSPH9" "PCDHGA4" "POLG" "PRICKLE2" "UBA2" "CEBPG" ......[127] "ZFHX4" "HERC2" "MYBBP1A" "SRRT" "COL1A2" "GPR155" "GPX2"[134] "IP6K2" "KRT86" "OR2T34" "SULT1A2" "ADGRD1" "PABPC1" "TBP"[141] "IGSF3"
KEGG注释
KEGG 注释用到的还是 Y叔 的 clusterProfiler,这里我是简单定义了一个函数 kegg_SYMBOL_hsa,后面需要用到就直接调动该函数就好。
library(org.Hs.eg.db)library(clusterProfiler)kegg_SYMBOL_hsa <- function(genes){gene.df <- bitr(genes, fromType = "SYMBOL",toType = c("SYMBOL", "ENTREZID"),OrgDb = org.Hs.eg.db)head(gene.df)diff.kk <- enrichKEGG(gene = gene.df$ENTREZID,organism = 'hsa',pvalueCutoff = 0.99,qvalueCutoff = 0.99)return( setReadable(diff.kk, OrgDb = org.Hs.eg.db,keyType = 'ENTREZID'))}
对于 trunk mutations,这些突变主要富集的通路是:
trunk_kk=kegg_SYMBOL_hsa(trunk_gene)trunk_df=trunk_kk@resultwrite.csv(trunk_df,file = 'trunk_kegg.csv')png(paste0('trunk_kegg', '.png'),width = 1080,height = 540)barplot(trunk_kk,font.size = 20)dev.off()
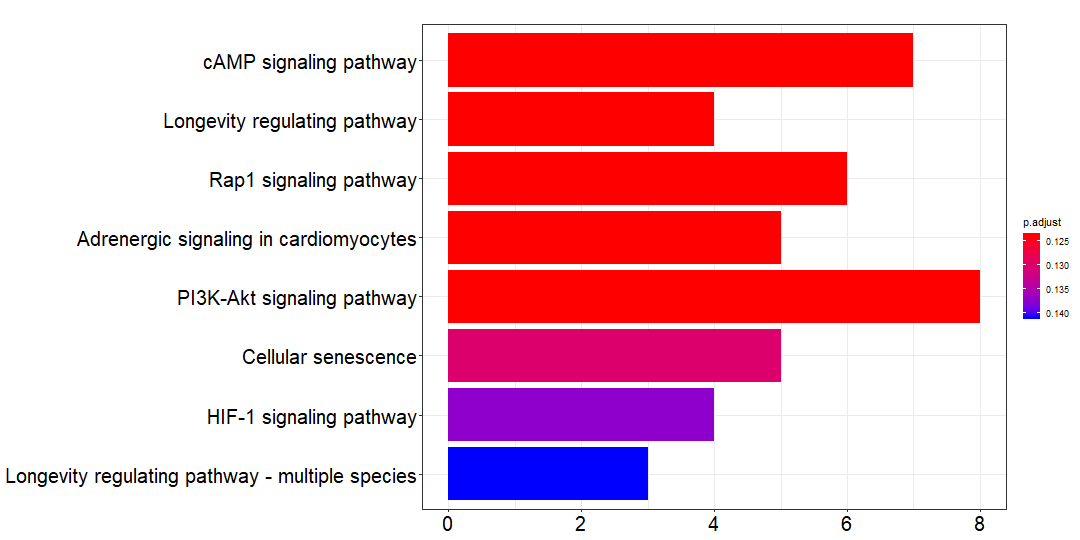
第一条通路是 cAMP signaling pathway:
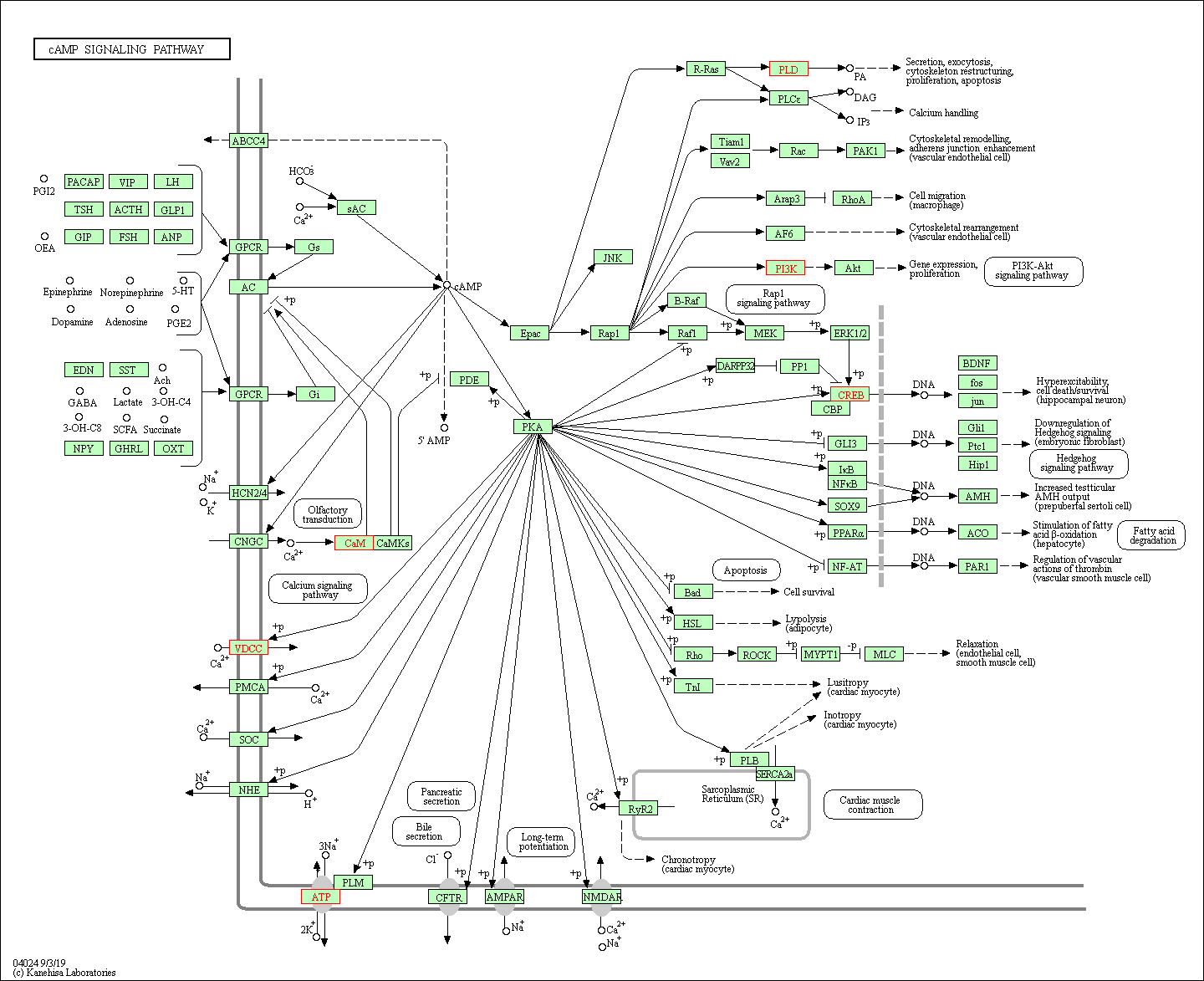
cAMP is one of the most common and universal second messengers, and its formation is promoted by adenylyl cyclase (AC) activation after ligation of G protein-coupled receptors (GPCRs) by ligands including hormones, neurotransmitters, and other signaling molecules. cAMP regulates pivotal physiologic processes including metabolism, secretion, calcium homeostasis, muscle contraction, cell fate, and gene transcription. cAMP acts directly on three main targets: protein kinase A (PKA), the exchange protein activated by cAMP (Epac), and cyclic nucleotide-gated ion channels (CNGCs). PKA modulates, via phosphorylation, a number of cellular substrates, including transcription factors, ion channels, transporters, exchangers, intracellular Ca2+ -handling proteins, and the contractile machinery. Epac proteins function as guanine nucleotide exchange factors (GEFs) for both Rap1 and Rap2. Various effector proteins, including adaptor proteins implicated in modulation of the actin cytoskeleton, regulators of G proteins of the Rho family, and phospholipases, relay signaling downstream from Rap.
可视化出来,就可以看到发生突变的基因在该通路中的位置:
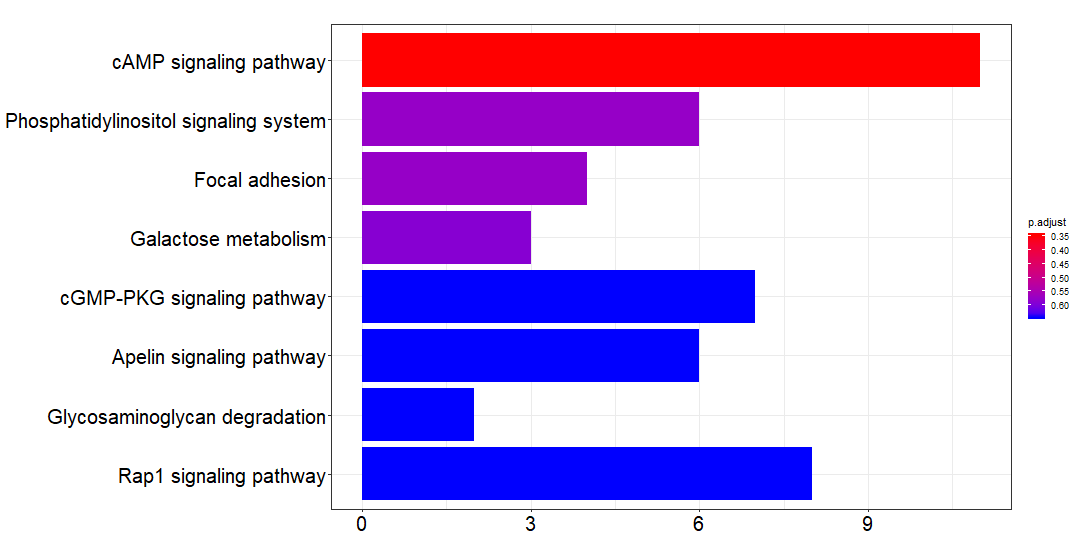
除此之外,还有 branch_gene 和 private_gene ,注释结果分别是:
branch_kk=kegg_SYMBOL_hsa(branch_gene)branch_df=branch_kk@resultwrite.csv(branch_df,file = 'branch_kegg.csv')png(paste0('branch_kegg', '.png'),width = 1080,height = 540)barplot(branch_kk,font.size = 20)dev.off()
private_kk=kegg_SYMBOL_hsa(private_gene)private_df=private_kk@resultwrite.csv(private_df,file = 'private_kegg.csv')png(paste0('private_kegg', '.png'),width = 1080,height = 540)barplot(private_kk,font.size = 20)dev.off()
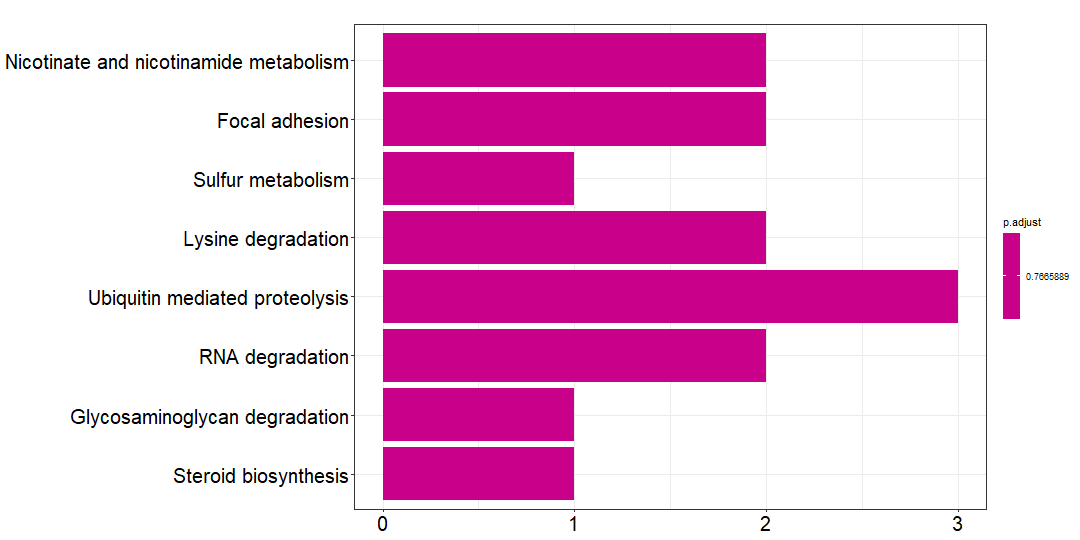
对比一下,private mutations 的注释结果基本上不显著,没有意义。但是 trunk 和 branch mutations 的注释结果还是比较满意的。

若有收获,就点个赞吧
0 人点赞
