fq 格式说明
从 sra 转格式后拿到的 fq 文件是测序原始的 fq ,记录了测序得到的 reads 的碱基序列和对应质量值,如case1_biorep_A_techrep_1.fastq.gz的前 8 行,以每 4 行为单位,作为一条 reads 的完整记录:
@case1_biorep_A_techrep.1/1 1 length=76AACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACC+case1_biorep_A_techrep.1/1 1 length=76A@?CDDABEDDDABDBBCAADBBCAADBCDABDCCC@BDBCCAADCCA@AECCCAAEDCDAAEDBDAAEDDA>=C=@case1_biorep_A_techrep.2/1 2 length=76GTTGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAGGGTTAG+case1_biorep_A_techrep.2/1 2 length=76@?=DDDA@AGEEA@@FCD??@FCD@AAGCC@@?GED@@@FCC=??FEE=@@DEE=??EDE=>;DDD@>>BBA><>>
其中第 4 行为质量值,由测序仪给出,根据 Phred 33 (Phred 64 基本淘汰)可以换算成实际的质量值
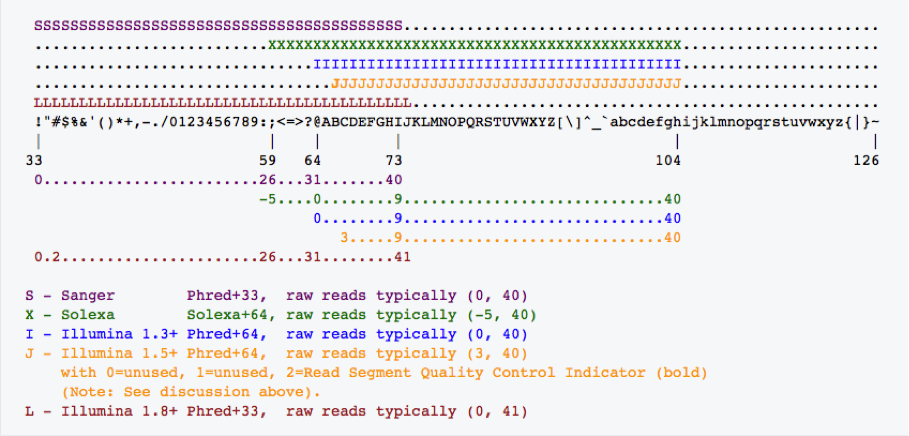
但是每一个 fq 文件中都有上千万条 reads ,这样换算显然是不科学的。
fastqc 的使用
前面我们已经安装了 fastqc 软件,接下来我们就用这个软件来对 fq 文件进行统计可视化,看看 reads 的读长、质量等分布情况。首先也是要激活环境,然后进入工作目录
conda activate wescd ~/wes_cancer/project
同样的通过 config 文件来进行批量操作,脚本如下:
cat config | while read iddofastqc --outdir ./3.qc/raw_qc/ --threads 16 ./1.raw_fq/${id}*.fastq.gz >> ./3.qc/raw_qc/${id}_fastqc.log 2>&1donemultiqc ./3.qc/raw_qc/*zip -o ./3.qc/raw_qc/multiqc
这里的参数--threads 16 指的是调用的服务器的 16 个线程来处理数据。
经过 fastqc 处理后,每个 fastq 文件会生成一个 html 文件和一个 zip 压缩文件,其中 html 文件是质控报告,可以下载到本地并用浏览器打开查看,而 zip 文件里面是 html 文件图表对应的数据。因为样本数较多,我们就用 multiqc 软件对所有样本的结果进行整合,方便一起查看。
质控和去接头
从上面的质控报告可以发现,有一些 reads 的质量值较差,或者测到了接头,这样的 reads 对于后续的分析是有一定的负面影响的,所以我们需要去除掉这些质量较差的 reads 或者切掉测到的接头,一般使用的软件是 trim-galore,软件会自动检测已知的常用接头。
## trim_galore.shcat config | while read iddofq1=./1.raw_fq/${id}_1.fastq.gzfq2=./1.raw_fq/${id}_2.fastq.gztrim_galore --paired -q 28 --phred33 --length 30 --stringency 3 --gzip --cores 8 -o ./2.clean_fq $fq1 $fq2 >> ./2.clean_fq/${id}_trim.log 2>&1donenohup bash trim_galore.sh &
trim 处理后的生成文件名带有 val 标签,感兴趣可以比较 trim 前后的文件大小,但是这里我们还是再做一次 fastqc ,通过质控报告来对比 trim 前后的结果
cat config | while read iddofastqc --outdir ./3.qc/clean_qc/ --threads 16 ./2.clean_fq/${id}*.fq.gz >> ./3.qc/clean_qc/${id}_fastqc.log 2>&1donemultiqc ./3.qc/clean_qc/*zip -o ./3.qc/clean_qc/multiqc

若有收获,就点个赞吧
0 人点赞
