写在前面
前面我们由 pyclone 分析了肿瘤样本的 clusters 结构,接下来我们进一步分析肿瘤进化,画一个鱼图,需要用到的工具是 citup 和 Timescape
参考:
https://github.com/sfu-compbio/citup
https://shahlab.ca/projects/citup/
https://www.cnblogs.com/xlij1205/p/10848917.html
https://bioconductor.org/packages/release/bioc/vignettes/timescape/inst/doc/timescape_vignette.html
citup 软件的安装和使用
CITUP全称 Conality Inference in Tumors Using Phylogeny,是一种使用系统发育论进行肿瘤的克隆推断生物信息学工具,可用于从单个患者获得的多个样本来推断肿瘤异质性。给定每个样本的突变频率,CITUP使用基于优化的算法来找到最能解释数据的进化树。但是,前提条件是,CITUP也是针对于深度测序数据的,这次勉强使用,为的是学习一下这个工具。
官网推荐我们使用 conda 来安装
# 添加channelsconda config --add channels http://conda.anaconda.org/dranewconda config --add channels https://conda.anaconda.org/IBMDecisionOptimization/linux-64# 创建小环境,然后安装,其实可以安装在前面的 pyclone 小环境里conda create -n citupconda activate citupconda install -y citup h5py# 安装完成后可以调用软件的帮助文档run_citup_iter.py -hrun_citup_qip.py -h
实际运行需要准备两个文件,一个是突变位点在不同样本的突变频率,行是突变位点,列是样本。另一个是突变的 cluster,只有单列,记录每个突变位点所在的 cluster 。两个文件的突变位点顺序要一致。具体可以参考:https://shahlab.ca/projects/citup/
在这里,我用前面 pyclone 结果进行处理:
for case in case{1..6}docat ./9.pyclone/${case}_pyclone_analysis/tables/loci.tsv | cut -f 6 | sed '1d' | paste - - - - >./9.pyclone/${case}_pyclone_analysis/freq.txtcat ./9.pyclone/${case}_pyclone_analysis/tables/loci.tsv | cut -f 3 | sed '1d' | paste - - - - |cut -f 1 >./9.pyclone/${case}_pyclone_analysis/cluster.txt## 获取 sample_id 信息,后面画图要用到cat ./9.pyclone/${case}_pyclone_analysis/tables/loci.tsv | cut -f 2 | sed '1d' | head -4 >./9.pyclone/${case}_pyclone_analysis/sample_iddone
之后得到的文件结构就是:
$ ls ./9.pyclone/*./9.pyclone/case1_pyclone_analysis:cluster.txt config.yaml freq.txt plots sample_id tables trace yaml./9.pyclone/case2_pyclone_analysis:cluster.txt config.yaml freq.txt plots sample_id tables trace yaml./9.pyclone/case3_pyclone_analysis:cluster.txt config.yaml freq.txt plots sample_id tables trace yaml./9.pyclone/case4_pyclone_analysis:cluster.txt config.yaml freq.txt plots sample_id tables trace yaml./9.pyclone/case5_pyclone_analysis:cluster.txt config.yaml freq.txt plots sample_id tables trace yaml./9.pyclone/case6_pyclone_analysis:cluster.txt config.yaml freq.txt plots sample_id tables trace yaml
每个样本的 3 个文件是(以 case1 为例):
$ cat ./9.pyclone/case1_pyclone_analysis/freq.txt0.33636363636363636 0.41732283464566927 0.35135135135135137 0.121621621621621630.2191780821917808 0.28440366972477066 0.2125 0.119047619047619040.31137724550898205 0.2519480519480519 0.2246153846153846 0.13624678663239073......$ cat ./9.pyclone/case1_pyclone_analysis/cluster.txt1000......$ cat ./9.pyclone/case1_pyclone_analysis/sample_idcase1_techrep_2case1_biorep_A_techrepcase1_biorep_Bcase1_biorep_C
然后就是运行,输入文件是 freq.txt 和 cluster.txt,结果输出到 reasults.h5:
for case in case{1..6}dorun_citup_qip.py --submit local \./9.pyclone/${case}_pyclone_analysis/freq.txt \./9.pyclone/${case}_pyclone_analysis/cluster.txt \./9.pyclone/${case}_pyclone_analysis/results.h5done
接下来需要用 python 处理一下结果,因为得到的结果results.h5是一个hdf5格式,不是普通的文本文件,处理起来比较麻烦一些,当然用 R 语言也是可以处理的,只不过这个文件太大了,有 30M+,所以才选择用 python:

然后就是从上述结果中取最佳拟合树、进化树节点,克隆组成等信息,我把处理的过程写成一个 python 脚本 citup.py:
import sysimport h5pyhf=h5py.File(sys.argv[1],'r')hf.keys()opnum=hf["results/optimal/index"][0]cellfreq=hf["trees/" + str(opnum) + "/clone_freq/block0_values"][:]tree=hf["trees/" + str(opnum) + "/adjacency_list/block0_values"][:]print cellfreqprint tree
然后就是批量处理,由于我的 python 基础太差,写出来的这个脚本有点丑:
for case in case{1..6}dopython citup.py ./9.pyclone/${case}_pyclone_analysis/results.h5 | sed 's/^ \[//;s/\[//g;s/\]//g' | tr ' ' '\t'| grep '\.' >./9.pyclone/${case}_pyclone_analysis/cellfreq.txtpython citup.py ./9.pyclone/${case}_pyclone_analysis/results.h5 | sed 's/^ \[//;s/\[//g;s/\]//g' | tr ' ' '\t'| grep -v '\.' >./9.pyclone/${case}_pyclone_analysis/tree.txtdone
得到的 cellfreq.txt 和 tree.txt 这两个文件,前者行表示样本,列表示克隆 clusters,每个值代表克隆频率。后者表示的是进化树的克隆分支关系
$ cat ./9.pyclone/case1_pyclone_analysis/cellfreq.txt0.35833 0.076023 0.235088 0.3305590.348454 0.0699815 0.235574 0.345990.401151 0.101147 0.217818 0.2798830.751012 0.00999059 0.120444 0.118553$ cat ./9.pyclone/case1_pyclone_analysis/tree.txt0 11 20 3
Timescape 可视化
得到上面的结果,就可以进一步处理成 timescape 的 input 了,处理的方法是:
The adjacency list can be written as a TSV with the column names
source,targetto be input into E-scape, and the clone frequencies should be reshaped such that each row represents a clonal frequency in a specific sample for a specific clone, with the columns representing the time or space ID, the clone ID, and the clonal prevalence.
也就是 tree.txt 不用做修改,cellfreq.txt 需要被处理为3列 time or space ID, the clone ID, and the clonal prevalence,R代码部分如下:
BiocManager::install("timescape")library(timescape)options(stringsAsFactors = F)example("timescape")browseVignettes("timescape")library(plotly)library(htmlwidgets)library(webshot)for (i in 1:6) {# i = 1# treetree_edges = read.table(paste0("9.pyclone/case", i, "_pyclone_analysis/tree.txt"))colnames(tree_edges) = c("source","target")# clonal prevalencescellfreq = read.table(paste0("9.pyclone/case", i, "_pyclone_analysis/cellfreq.txt"))colnames(cellfreq) = 0:(length(cellfreq)-1)sample_id = read.table(paste0("9.pyclone/case", i, "_pyclone_analysis/sample_id"))cellfreq$timepoint = sample_id[ , 1]library(tidyr)clonal_prev = gather(cellfreq, key="clone_id", value = "clonal_prev", -timepoint)clonal_prev = clonal_prev[order(clonal_prev$timepoint),]# targeted mutations# mutations <- read.csv(system.file("extdata", "AML_mutations.csv", package = "timescape"))p = timescape(clonal_prev = clonal_prev, tree_edges = tree_edges,height=260)saveWidget(p, paste0("case", i,"_timescape", ".html"))}
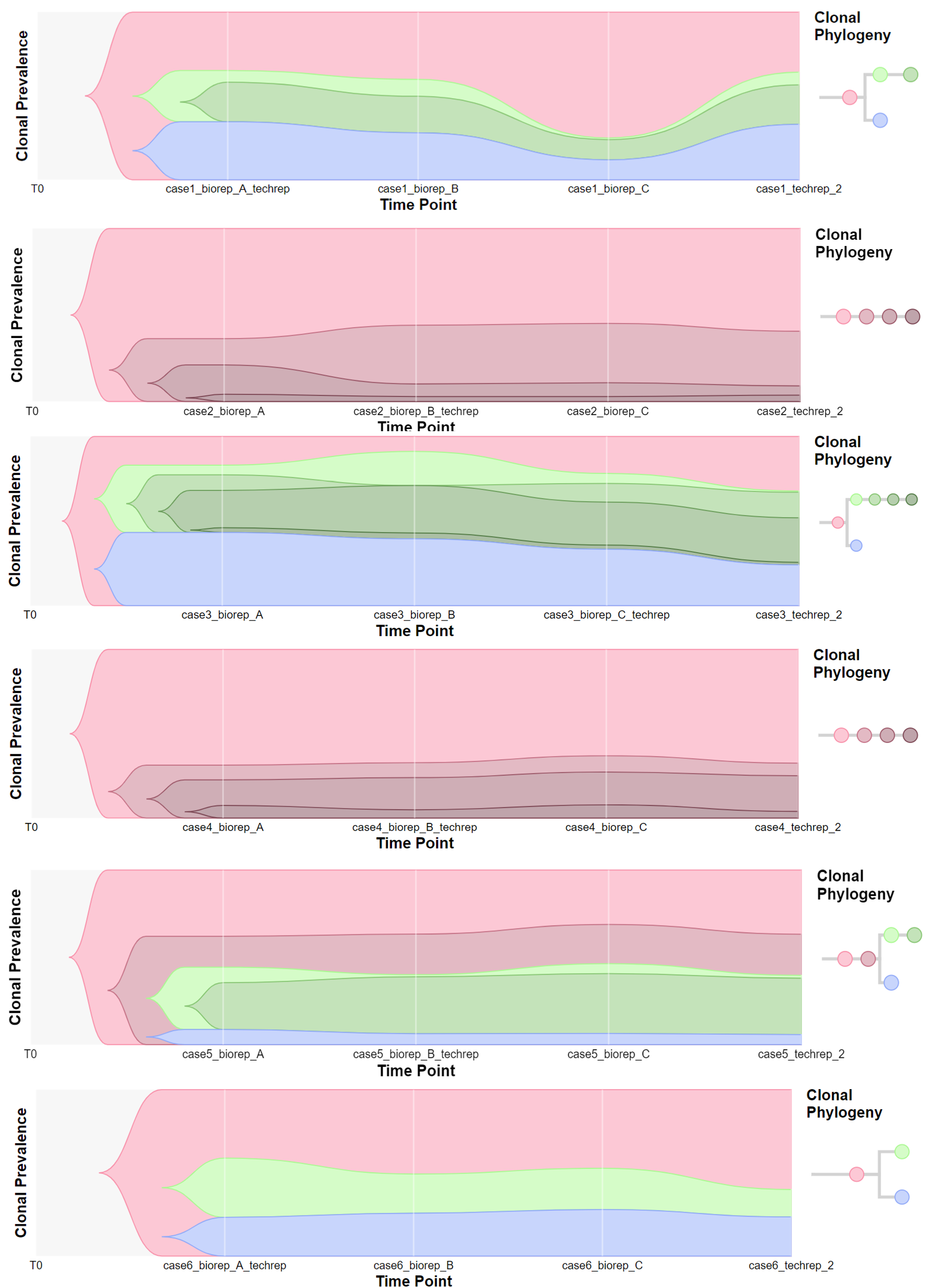
因为 timescape 的输出格式比较少见,上面我只能保存为网页文件,原文件是动态交互的,找不到合适的转为图片的方法,所以下面我就直接放了截图。可以看到,每个样本的克隆进化关系如图所示,当然结果并不是很理想,毕竟数据不是很完美,测序深度不是很符合要求,不过这并不影响我们对工具的学习:

若有收获,就点个赞吧
0 人点赞
