CNVkit
CNVkit 的用法比较简单,可以参考官网的教程:https://cnvkit.readthedocs.io/en/stable/index.html,写得很详细,这里我不再说明。下面是我用到的脚本(在这里我比较关心的是最后拿到的 segment 文件,即 final.seg,该文件可以用直接载入到 IGV 进行可视化,当然也可以用 cnvkit.py export 生成这样的 segments 文件,两者基本一样,然后和上一节 GATK 的结果进行比较)
## cnvkit.shGENOME=~/wes_cancer/data/Homo_sapiens_assembly38.fastabed=~/wes_cancer/data/hg38.exon.bedcnvkit.py batch ./5.gatk/*[ABC]*bqsr.bam ./5.gatk/*techrep_2_bqsr.bam \--normal ./5.gatk/*germline_bqsr.bam \--targets ${bed} \--fasta $GENOME \--drop-low-coverage --scatter --diagram --method amplicon \--output-reference my_reference.cnn --output-dir ./8.cnv/cnvkitcd ./8.cnv/cnvkitcnvkit.py export seg *bqsr.cns -o gistic.segmentssed 's/_bqsr//' gistic.segmentsawk '{print FILENAME"\t"$0}' *bqsr.cns | grep -v chromosome |sed 's/_bqsr.cns//g' |awk '{print $1"\t"$2"\t"$3"\t"$4"\t"$8"\t"$6}' >final.seg
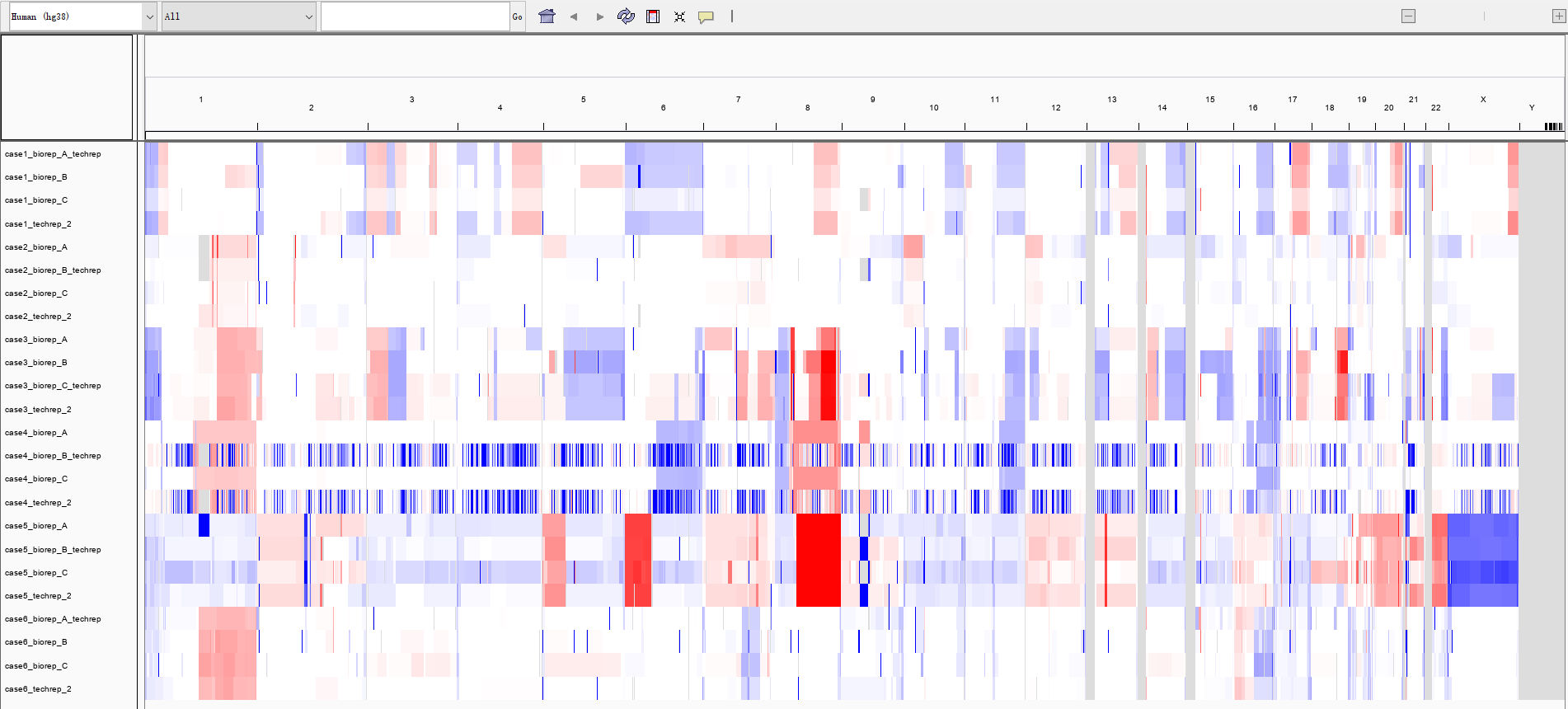
下面两张图是 CNVkit 结果和上一节 GATK CNV 流程的结果,对比两个图,发现这两个软件拿到的结果基本一样,同样的 case4 技术重复的两个样本拷贝数缺失事件碎片化。

GISTIC2
GISTIC2 的安装比较复杂,需要安装 MATLAB,具体可以参考曾老师的博客:用 GISTIC 多个 segment 文件来找SCNA 变异 http://www.bio-info-trainee.com/1648.html
cd ~/wes_cancer/project/8.cnv/gistic## 下载解压wget -c ftp://ftp.broadinstitute.org/pub/GISTIC2.0/GISTIC_2_0_23.tar.gztar zvxf GISTIC_2_0_23.tar.gz## 安装MATLABcd MCR_Installer/unzip MCRInstaller.zip./install -mode silent -agreeToLicense yes -destinationFolder ~/wes_cancer/project/8.cnv/gistic/MATLAB_Compiler_Runtime/## 安装后的文件结构## 添加环境变量echo "export XAPPLRESDIR=~/wes_cancer/project/8.cnv/gistic/MATLAB_Compiler_Runtime/v83/X11/app-defaults:\$XAPPLRESDIR" >> ~/.bashrcsource ~/.bashrc
安装完成之后,新建一个目录,把上一步 cnvkit 拿到的 gistic.segments 文件拷贝到当前目录
mkdir SRP070662_gisticcp ~/wes_cancer/project/8.cnv/cnvkit/gistic.segments ./SRP070662_gistic
然后把将脚本 run_gistic_example 做了一定的修改,实际运行 GISTIC 的脚本 my.gistic.sh 如下:
#!/bin/sh## run example GISTIC analysis## output directoryecho --- creating output directory ---basedir=`pwd`/SRP070662_gistic# mkdir -p $basedirecho --- running GISTIC ---## input file definitionssegfile=`pwd`/SRP070662_gistic/gistic.segments#markersfile=`pwd`/examplefiles/markersfile.txtrefgenefile=`pwd`/refgenefiles/hg38.UCSC.add_miR.160920.refgene.mat#alf=`pwd`/examplefiles/arraylistfile.txt#cnvfile=`pwd`/examplefiles/cnvfile.txt## call script that sets MCR environment and calls GISTIC executable./gistic2 -b $basedir -seg $segfile -refgene $refgenefile -genegistic 1 -smallmem 1 -broad 1 -brlen 0.5 -conf 0.90 -armpeel 1 -savegene 1 -gcm extreme
生成的文件有
all_data_by_genes.txtall_lesions.conf_90.txtall_thresholded.by_genes.txtamp_genes.conf_90.txtamp_qplot.pdfamp_qplot.pngbroad_data_by_genes.txtbroad_significance_results.txtbroad_values_by_arm.txtD.cap1.5.matdel_genes.conf_90.txtdel_qplot.pdfdel_qplot.pngfocal_dat.0.5.matfocal_data_by_genes.txtfreqarms_vs_ngenes.pdfgistic_inputs.matpeak_regs.matperm_ads.matraw_copy_number.pdfraw_copy_number.pngregions_track.conf_90.bedsample_cutoffs.txtsample_seg_counts.txtscores.0.5.matscores.gisticwide_peak_regs.mat
可视化结果生成的 3 张图,3张图均未展示性染色体的拷贝数变异信息,图 1 垂直方向是基因组,水平方向是样本,如果倒过来,就和前面的 cnvkit 结果基本一致。图 2,3 表示的是拷贝数增加或者缺失的情况,水平方向下面是q值,绿线代表显着性阈值(q 值 = 0.25)。图上面是 G-score,某个区域的 G-score 分数越高,该区域的发生拷贝数变异事件的可能性就越大,偶然性越低。可以在输出文件 amp_genes.conf_90.txt和del_genes.conf_90.txt查看每一个区域所包含的基因。
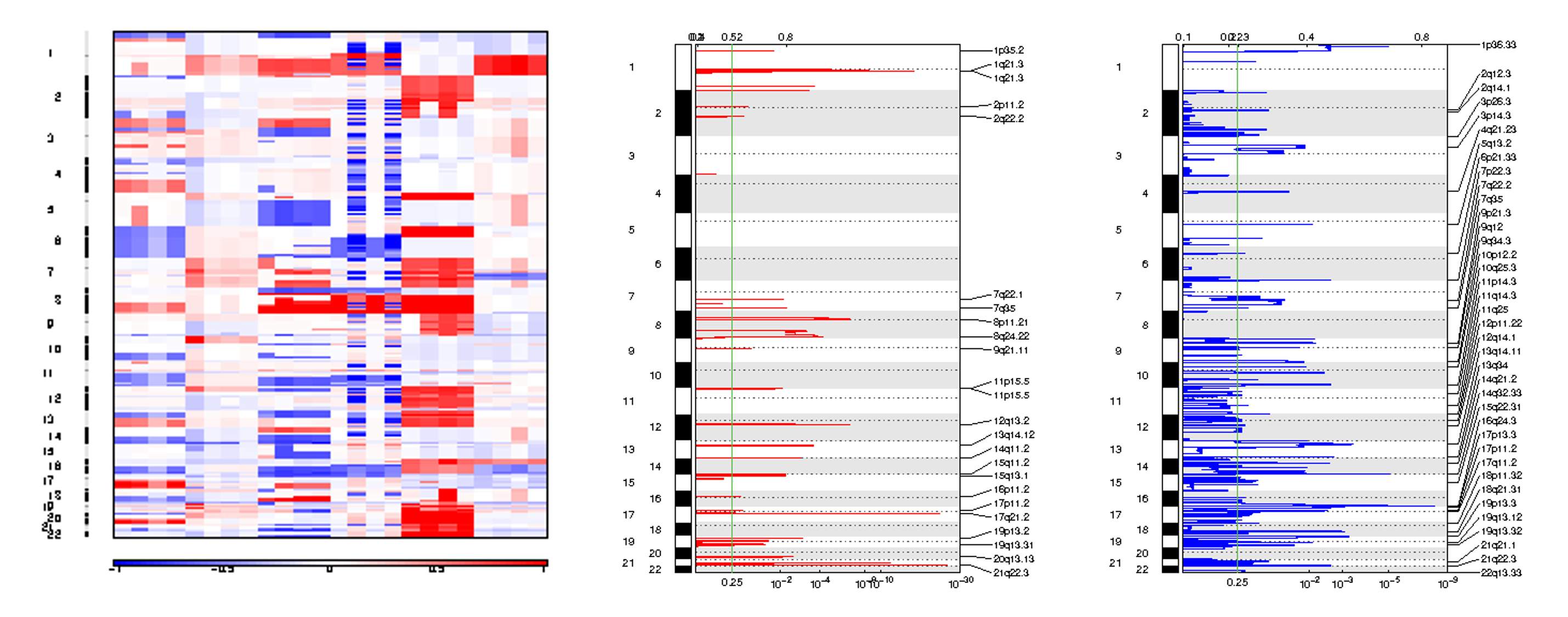
在这里,拷贝数缺失事件明显比拷贝数增加事件要多,可能是 case4 技术重复异常带来的影响,我试着删除掉 case4 技术重复的两个样本,对比一下结果(如下)发现,拷贝数增加和缺失事件分布: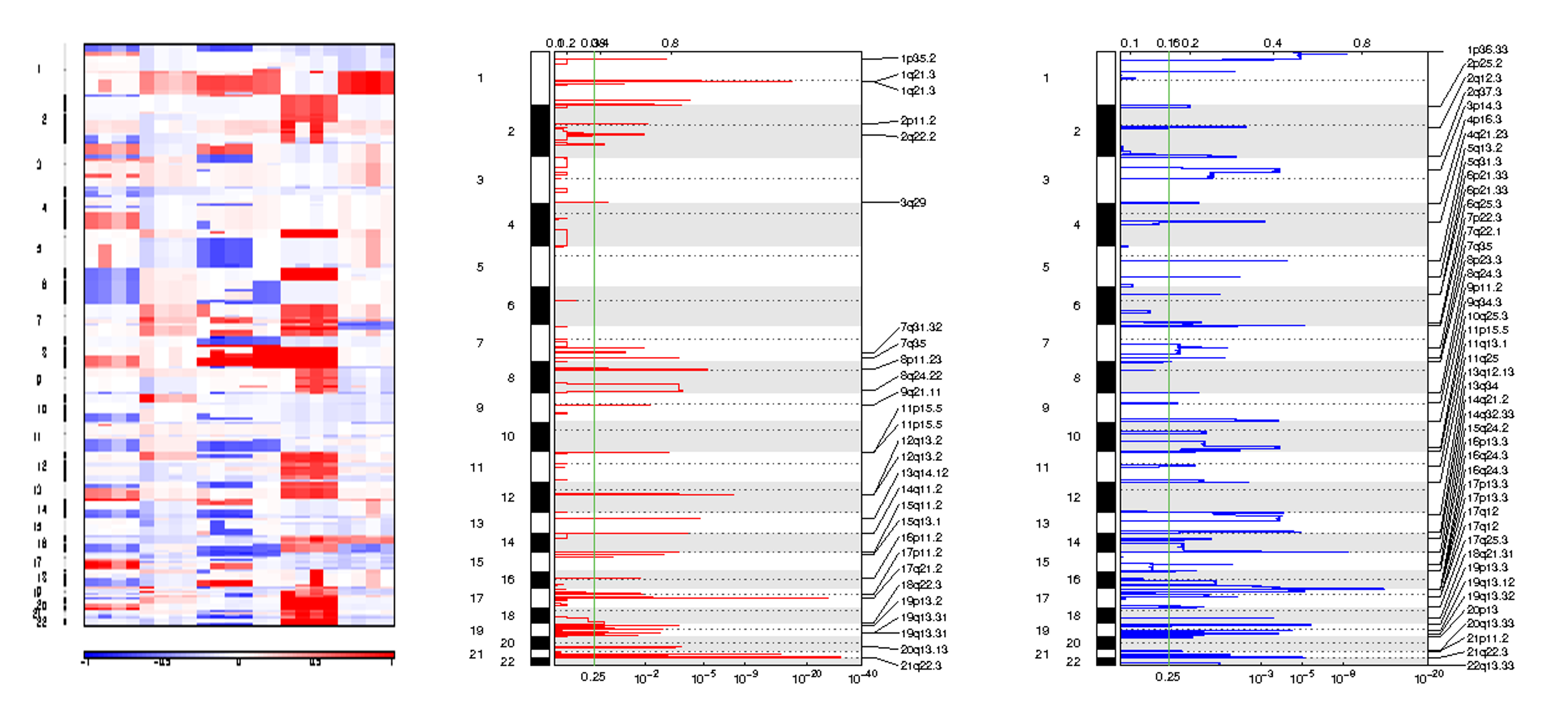
facet
facet是一个 R 包,但不在 CRAN 中收录,安装的时候有点麻烦,在 Mac 和 Linux 上安装没问题,在 windows 上安装没成功。 facet 的输入文件是处理后的 vcf 文件,在这里我用的是 GATK 的HaplotypeCaller工具生成的结果并走了gvcf流程拿到的 merge.vcf,最后载入 R 中进行分析, R 语言分析部分的代码如下:
if(F){install.packages('devtools')devtools::install_github("mskcc/facets", build_vignettes = TRUE)devtools::install_github("mskcc/pctGCdata")}options( stringsAsFactors = F )library("facets")# check if .Random.seed existsseedexists <- exists(".Random.seed")# save seedif(seedexists)oldSeed <- .Random.seed# Alway use the same random seedset.seed(0xfade)# read the dataif(F){library(vcfR)vcf_file='./5.gatk/gvcf/merge.vcf'### 直接读取群体gvcf文件即可vcf <- read.vcfR( vcf_file, verbose = FALSE )save(vcf,file = 'input_vcf.Rdata')}load(file = 'input_vcf.Rdata')vcf@fix[1:4,1:4]vcf@gt[1:14,1:4]colnames(vcf@gt)library(stringr)tmp_f <- function(x){#x=vcf@gt[,2]gt=str_split(x,':',simplify = T)[,1]ad=str_split(str_split(x,':',simplify = T)[,2],',',simplify = T)[,2]dp=str_split(x,':',simplify = T)[,3]return(data.frame(gt=gt,dp=as.numeric(dp),ad=as.numeric(ad)))}colnames(vcf@gt)tms=colnames(vcf@gt)[!grepl('_germline',colnames(vcf@gt))][-1]tmp <- lapply(tms, function(tm){#tm=tms[1]print(tm)nm=paste0(strsplit(tm,'_')[[1]][1],'_germline')print(nm)if(nm %in% colnames(vcf@gt)){snpm=cbind(vcf@fix[,1:2],tmp_f(vcf@gt[,nm]),tmp_f(vcf@gt[,tm]))## only keep tumor or normal have mutationskp=snpm[,3] %in% c('1/1','0/1') | snpm[,6] %in% c('1/1','0/1')table(kp)snpm=snpm[kp,]## remove those show ./. positionskp=snpm[,3] == './.' | snpm[,6]== './.'print(table(!kp))snpm=snpm[!kp,c(1,2,4,5,7,8)]rcmat=snpmrcmat$POS=as.numeric(rcmat$POS)dim(rcmat)rcmat=na.omit(rcmat)colnames(rcmat)=c("Chromosome", "Position","NOR.DP","NOR.RD","TUM.DP","TUM.RD")rcmat[,1]=gsub('chr','',rcmat$Chrom)## fit segmentation treexx = preProcSample(rcmat)## estimate allele specific copy numbersoo=procSample(xx,cval=150)oo$dipLogR## EM fit version 1fit=emcncf(oo)tmp=fit$cncfwrite.table(tmp,file = paste0('facets_cnv_',tm,'.seg.txt'),row.names = F,col.names = F,quote = F)head(fit$cncf)fit$purityfit$ploidypng(paste0('facets_cnv_',tm,'.png'),res=150,width = 880,height = 880)plotSample(x=oo,emfit=fit, sname=tm)dev.off()if(F){fit2=emcncf2(oo)head(fit2$cncf)fit2$purityfit2$ploidypng(paste0('facets_cnv2_',tm,'.png'),res=150,width = 880,height = 880)plotSample(x=oo,emfit=fit2, sname=tm)dev.off()}return(c( fit$dipLogR,fit$ploidy,fit$purity))}})tmp <- do.call(rbind,tmp)rownames(tmp)=tmscolnames(tmp)=c('dipLogR', 'ploidy', 'purity')write.csv(tmp,'ploidy_and_purity.csv')
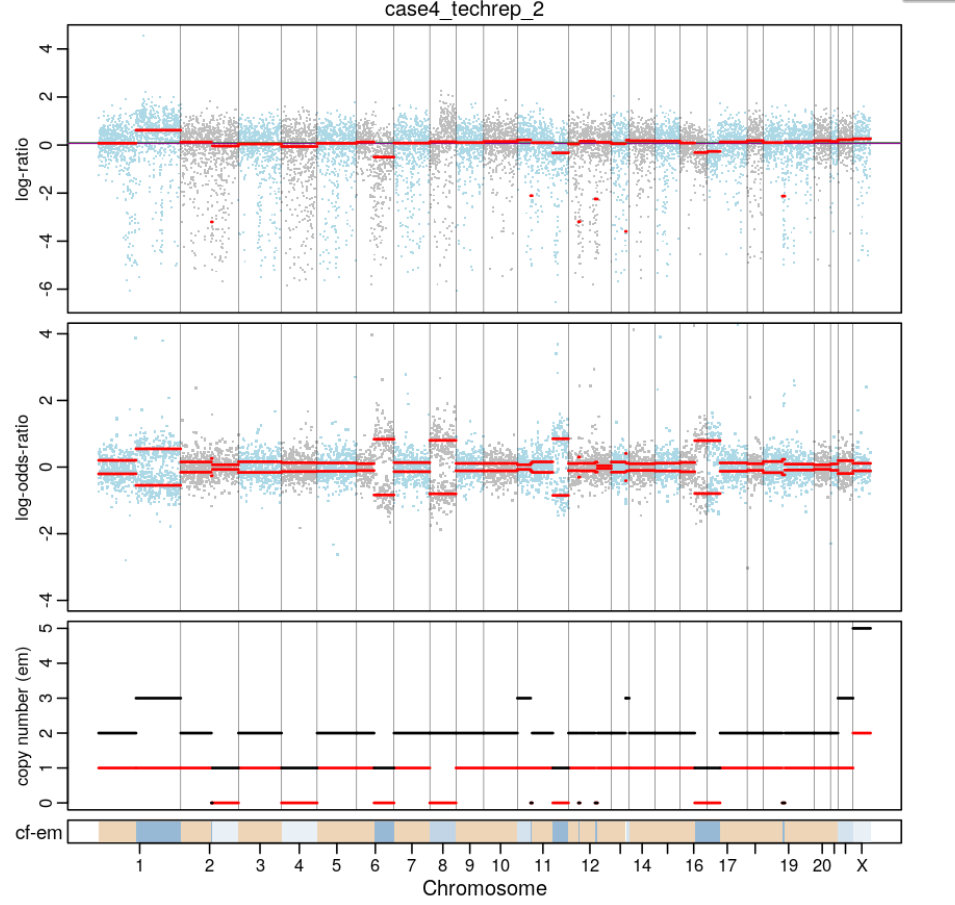
我们就拿case1_biorep_A_techrep样本的结果进行展示,主要关心图的第三个 panel ,绘制每个 segment 的主要(黑色 =2 )和次要(红色 = 1)拷贝数。底部栏显示相关的细胞分数(cf)。深蓝色表示高 cf。浅蓝色表示低 cf。米色表示正常。这个结果就和上面略有不同了,比如这个样本的 19 号染色体在第三个 panel(第一、二个panel 好像还是正常的)中显示是拷贝数增加,但在上面几个软件的结果中 19 号染色体的拷贝数均没有太大的变化。毕竟 facet 的输入数据是 germline mutations,和前面即个软件都不太一样。
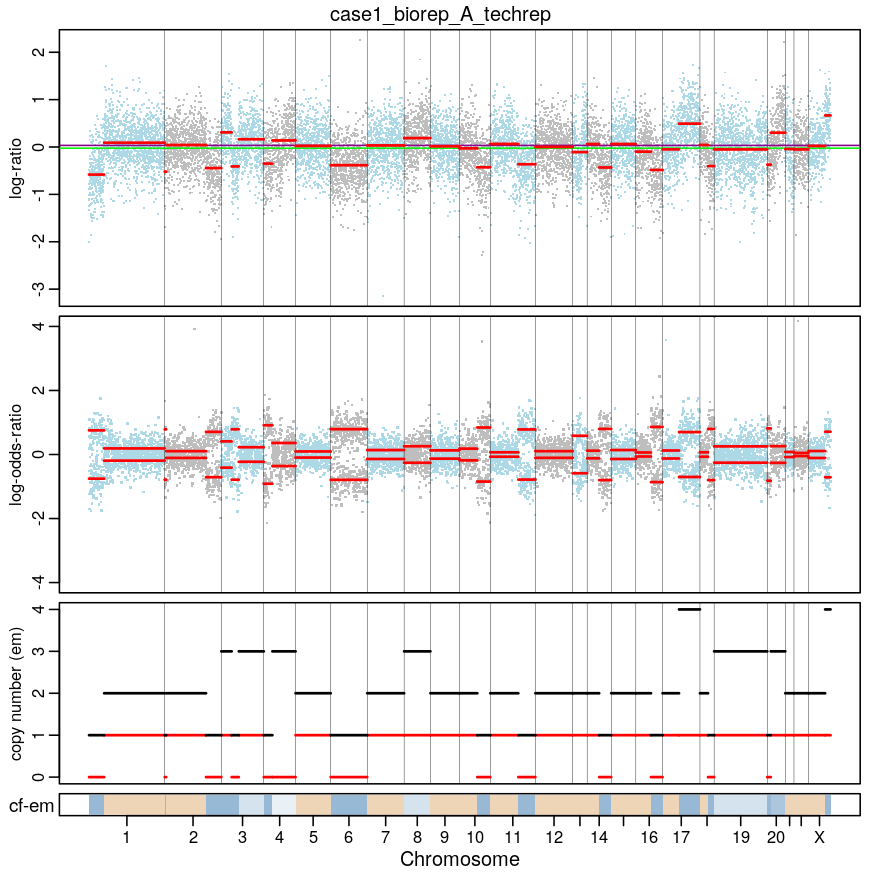
可能是软件基于不同类型的输入数据,算法不一样,facet 对 case4 技术重复的结果就比较正常了

若有收获,就点个赞吧
0 人点赞
