参考基因组及索引
通过 trim 过滤后的到的 fq,需要比对到参考基因组上才能让这些数据有意义。前面我们已经下载了人类参考基因组 hg38 版本,而且是从 GATK 数据库下载的,原文件大小为 800 多M,解压后为 3G 左右。同时也需要构建该参考基因组的 bwa 索引文件,即:
20K gatk_hg38.amb3.0G gatk_hg38.bwt1.5G gatk_hg38.sa445K gatk_hg38.ann768M gatk_hg38.pac
索引文件的作用类似于一本书的目录的作用,起到一个快速查找的功能。构建过程非常消耗时间和服务器的资源,方法是:
conda activate wescd ~/wes_cancer/datagunzip Homo_sapiens_assembly38.fasta.gztime bwa index -a bwtsw -p gatk_hg38 ~/wes_cancer/data/Homo_sapiens_assembly38.fastacd ~/wes_cancer/project
构建过程中会产生很多信息:
[bwa_index] Pack FASTA... 14.70 sec[bwa_index] Construct BWT for the packed sequence...[BWTIncCreate] textLength=6434693834, availableWord=464768632[BWTIncConstructFromPacked] 10 iterations done. 99999994 characters processed.[BWTIncConstructFromPacked] 20 iterations done. 199999994 characters processed.[BWTIncConstructFromPacked] 30 iterations done. 299999994 characters processed.[BWTIncConstructFromPacked] 40 iterations done. 399999994 characters processed.[BWTIncConstructFromPacked] 50 iterations done. 499999994 characters processed.......real 41m18.206suser 40m47.192ssys 2m30.302s
比对
有了索引文件之后,就可以开始比对,用到的比对软件是 bwa ,比对的 bwa.sh 脚本如下:
## bwa.shINDEX=~/wes_cancer/data/gatk_hg38cat config | while read iddoecho "start bwa for ${id}" `date`fq1=./2.clean_fq/${id}_1_val_1.fq.gzfq2=./2.clean_fq/${id}_2_val_2.fq.gzbwa mem -M -t 16 -R "@RG\tID:${id}\tSM:${id}\tLB:WXS\tPL:Illumina" ${INDEX} ${fq1} ${fq2} | samtools sort -@ 10 -m 1G -o ./4.align/${id}.bam -echo "end bwa for ${id}" `date`done
bwa 比对后先产生 sam 文件,但是因为 sam 文件太大,占用空间,要转换成二进制的 bam 文件,两者记录的信息是一样的。因此,通常都是把比对的结果通过管道符 | 传给 samtools 进行排序后直接生成 bam 文件(注意最后还有一个中划线 “-”作为缓存占位符),排序的依据是 bam 文件中 reads 比对到参考基因组的 染色体 和 坐标。生成的 bam 文件大小为 3 ~ 13G 不等。
13G case1_biorep_C.bam11G case1_biorep_B.bam12G case1_biorep_A_techrep.bam......3.4G case2_germline.bam5.0G case4_germline.bam7.4G case5_germline.bam
bam文件格式
bam(或者是 sam,cram )文件,是比对后拿到的文件,sam 文件是纯文本,非常占用存储空间,bam 是 sam 的二进制格式,cram 则是进一步压缩后的格式,这三者所记录的内容是一致的,但是 bam 文件是最常用的。记录了每一条 reads 比对到参考基因组的结果,主要有 11 列比较重要的信息(每一列以制表符分开):
| 列 | 列名 | 简介 | 举例 |
|---|---|---|---|
| 1 | QNAME | reads的名称 | case1_biorep_A_techrep.89335031 |
| 2 | FLAG | 由二进制数表示,每一个数字代表一种比对情况,这里的值是符合情况的数字相加总和,如75=1+2 1 +2 3 +2 6 |
129 |
| 3 | RNAME | 参考序列,一般是染色体 | chr17 |
| 4 | POS | 比对到染色体位置 | 42852401 |
| 5 | MAPQ | mapping质量 | 60 |
| 6 | CIGAR | MIDNSHP=XB这10个字符代表不同的含义,M比对成功,I插入,D删除 | 76M3D |
| 7 | RNEXT | 配对reads比对到的参考序列(染色体) | chr11 |
| 8 | PNEXT | 配对reads比对到染色体的位置 | 96427099 |
| 9 | TLEN | 可以理解为测序是文库插入片段长度 | 0 |
| 10 | SEQ | 序列,fq文件的第二行 | GCTTC…CCAGC |
| 11 | QUAL | 质量值,fq文件第四行 | @@@?D…CA@DD |
更多的说明可以查看官方教程:https://samtools.github.io/hts-specs/SAMv1.pdf
提取小bam
由于 bam 文件特别大,下载到本地载入 IGV 非常耗资源,所以我们就提取一个小 bam 进行演示。首先,要构建索引,提取小 bam,这里我们提取 17 号染色体的信息:chr17。再对小 bam 文件构建索引,因为 IGV 要求 bam 文件需要带索引才能载入,以样本 case1_biorep_A_techrep 为例。
samtools index case1_biorep_A_techrep.bamsamtools view -h case1_biorep_A_techrep.bam chr17 | samtools view -Sb - > small.bamsamtools index small.bam
对比一下大小,如果还觉得太大,上面提取的参数chr17可以改为类似 chr17:42850000-42950000,即提取特定坐标的部分。
12G case1_biorep_A_techrep.bam
694M small.bam
载入IGV
然后把小 bam 文件及其索引下载到本地,打开 IGV,加载 hg38 参考基因组,然后把 bam 文件拖到 IGV 窗口中
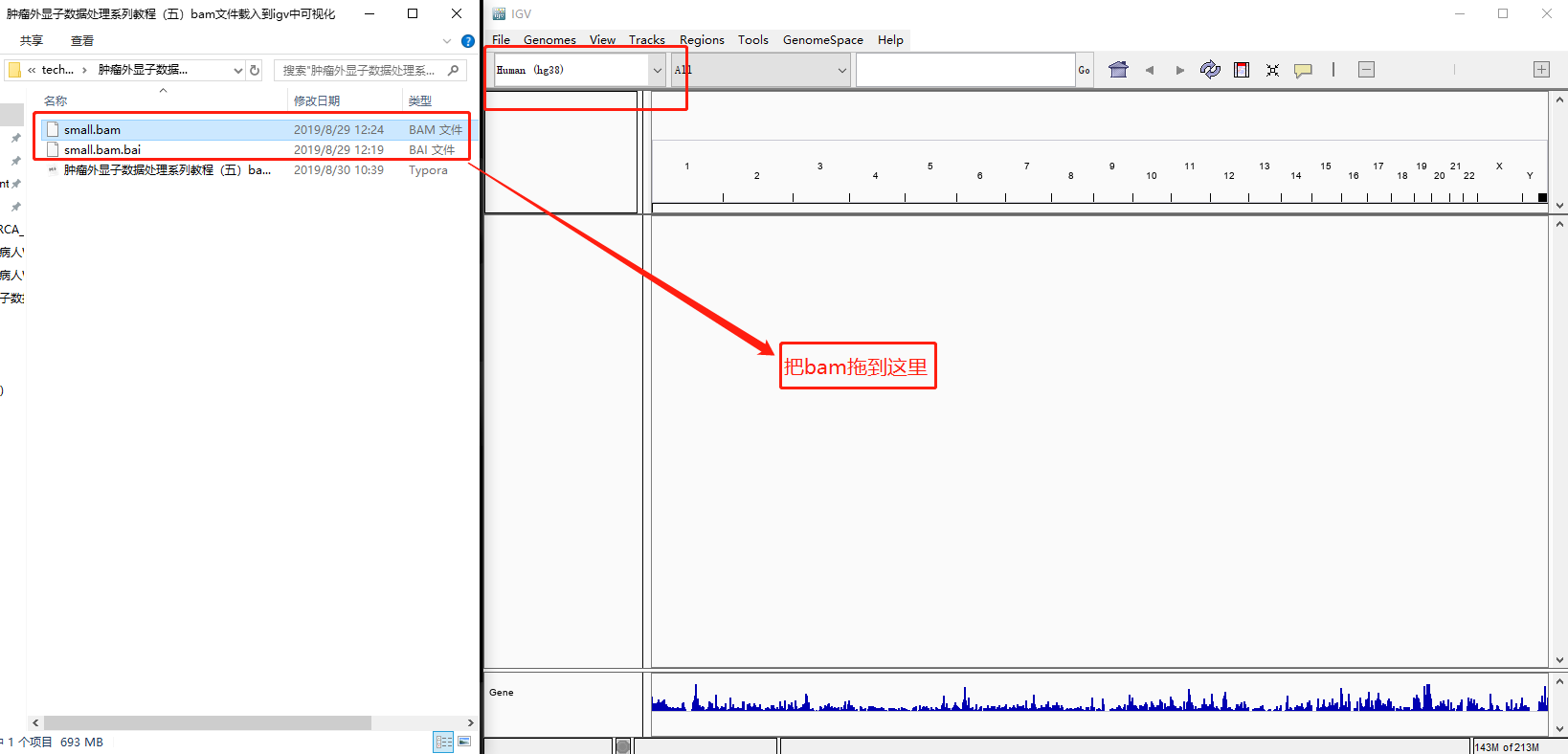
点击17和右上角的+
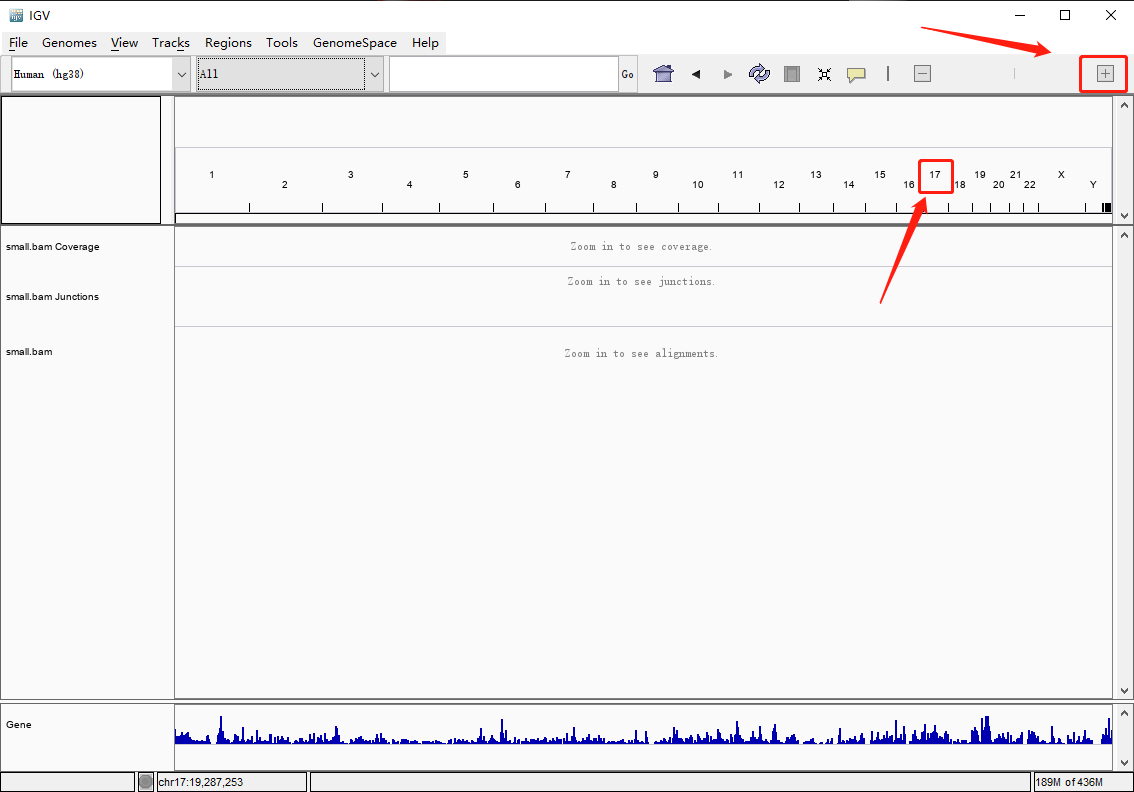
直到标尺显示的尺度小于 30kb( IGV 默认小于 30kb 才会显示 reads),就可以看到 reads 的信息
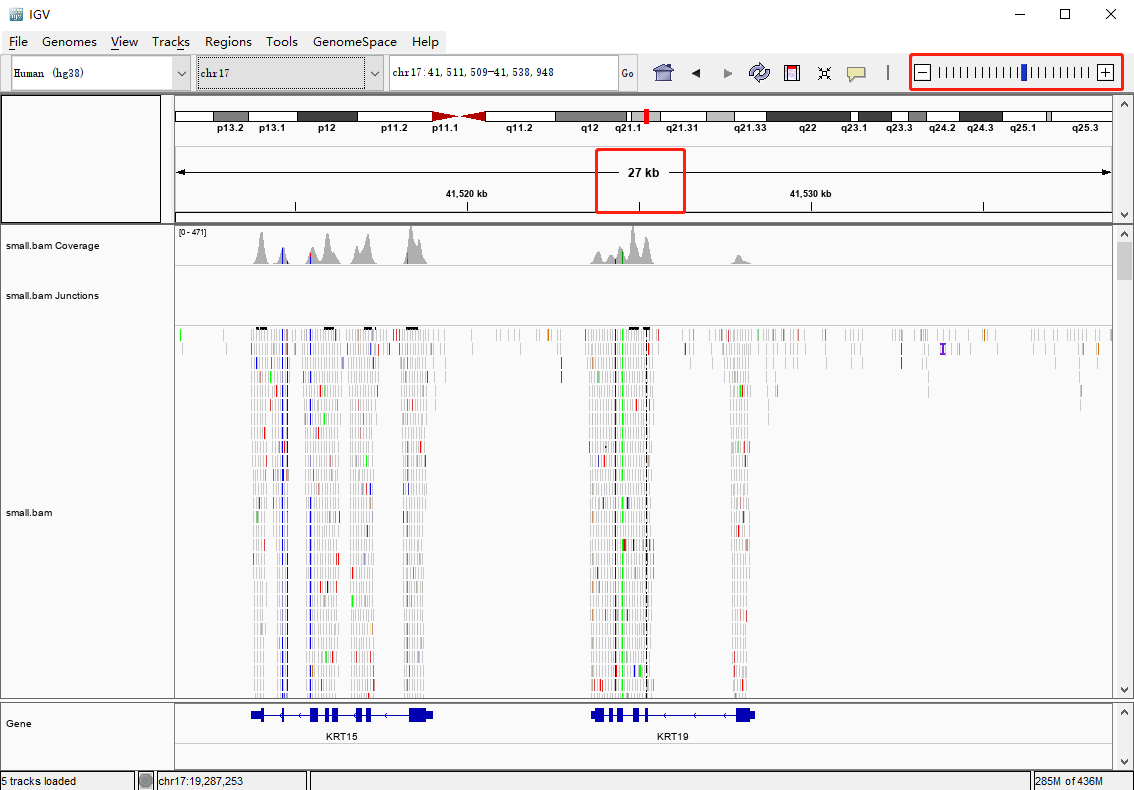
定位到感兴趣的位置
也可以在搜索框中输入感兴趣的位置或者基因,比如chr17:42,850,644-42,853,784
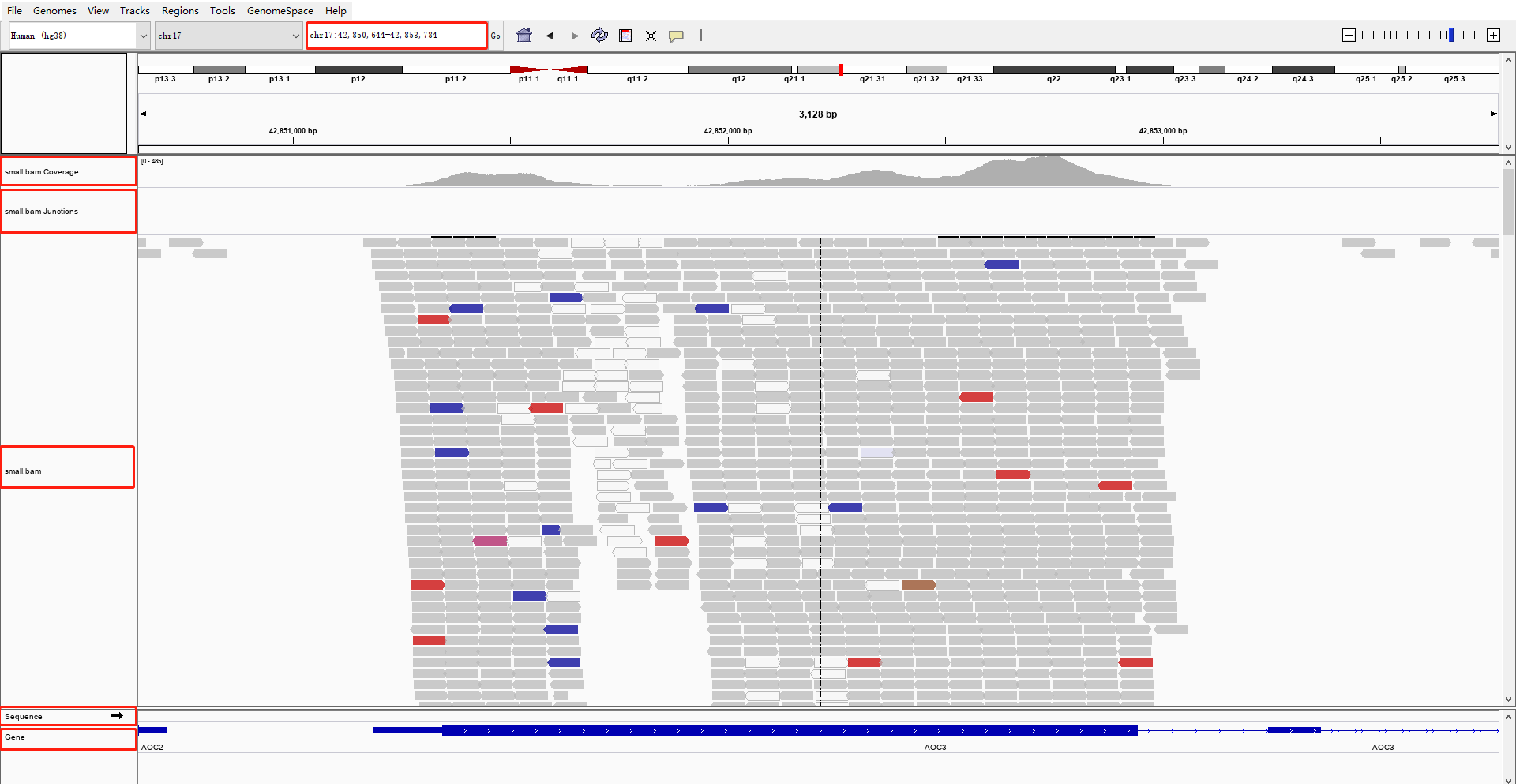
这里我们可以看到有5条 track:
- 第一条是 Coverage 即覆盖度,可以直观的看出染色体的每一处 reads 的覆盖情况,因为我们是外显子捕获测序,所以极大部分的 reads 都覆盖到了参考基因组的外显子区域及其侧翼区域。值得注意的是在这一行开头有峰的高度范围,且默认是动态变化的。
- 第二条是 junctions,一般用于转录组数据,可以看可变剪切,默认是不显示的,这里是我之前设置。
- 第三条是 bam 文件中reads详细信息,每一块代表一条 reads,将鼠标放到某一条 reads 上,就会呈现该 reads 的详细比对信息,与 bam 文件的中信息相对应。而且可以通过鼠标右键,调出设置菜单,方便进一步探索。
- 第四条是 sequence,参考基因组的序列碱基信息,当放大至一定程度时就会显示出来。
- 第五条是参考基因组注释信息,可以看到有基因、外显子、内含子、5’UTR、3’UTR 等,还可以右键选择显示基因的各个转录本。
颜色代表的含义
可以看到,这些 reads 大多数是灰色的,部分是透明,少部分是红色、蓝色、棕色等等,不同的颜色有不同的含义。
灰色:指的是比对成功,没有其他特别的含义
透明:指的是比对失败,对应的是 bam 文件中第 5 列,MAPQ 比对质量值,我们把鼠标放到透明的 reads 上就可以看到
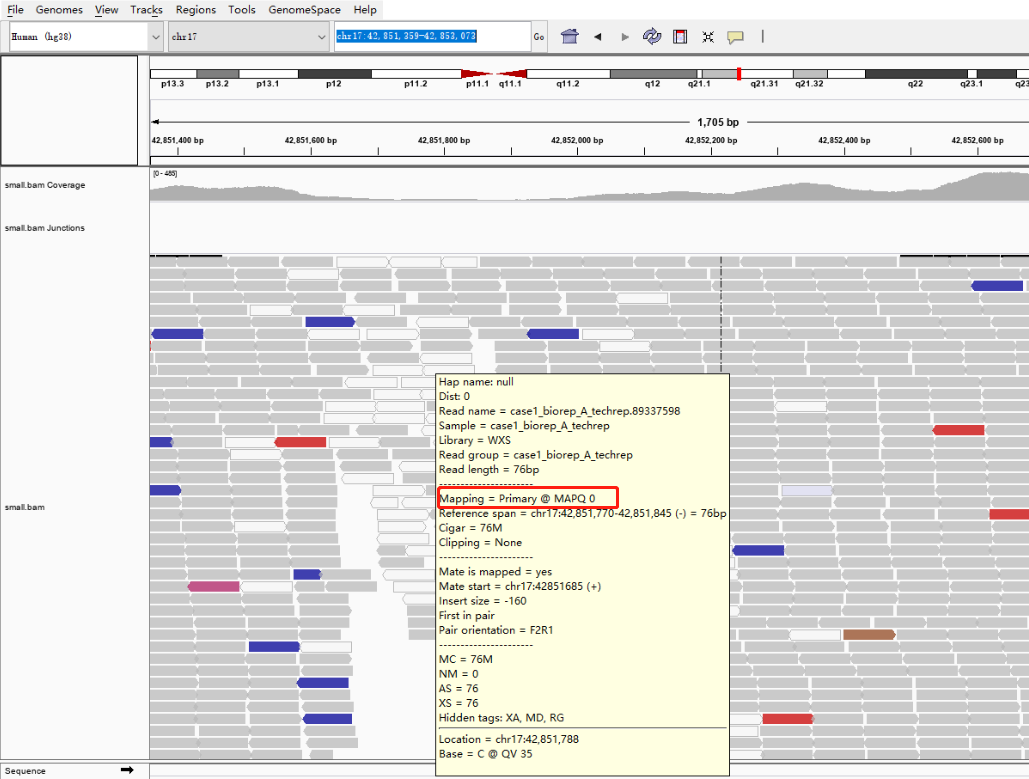
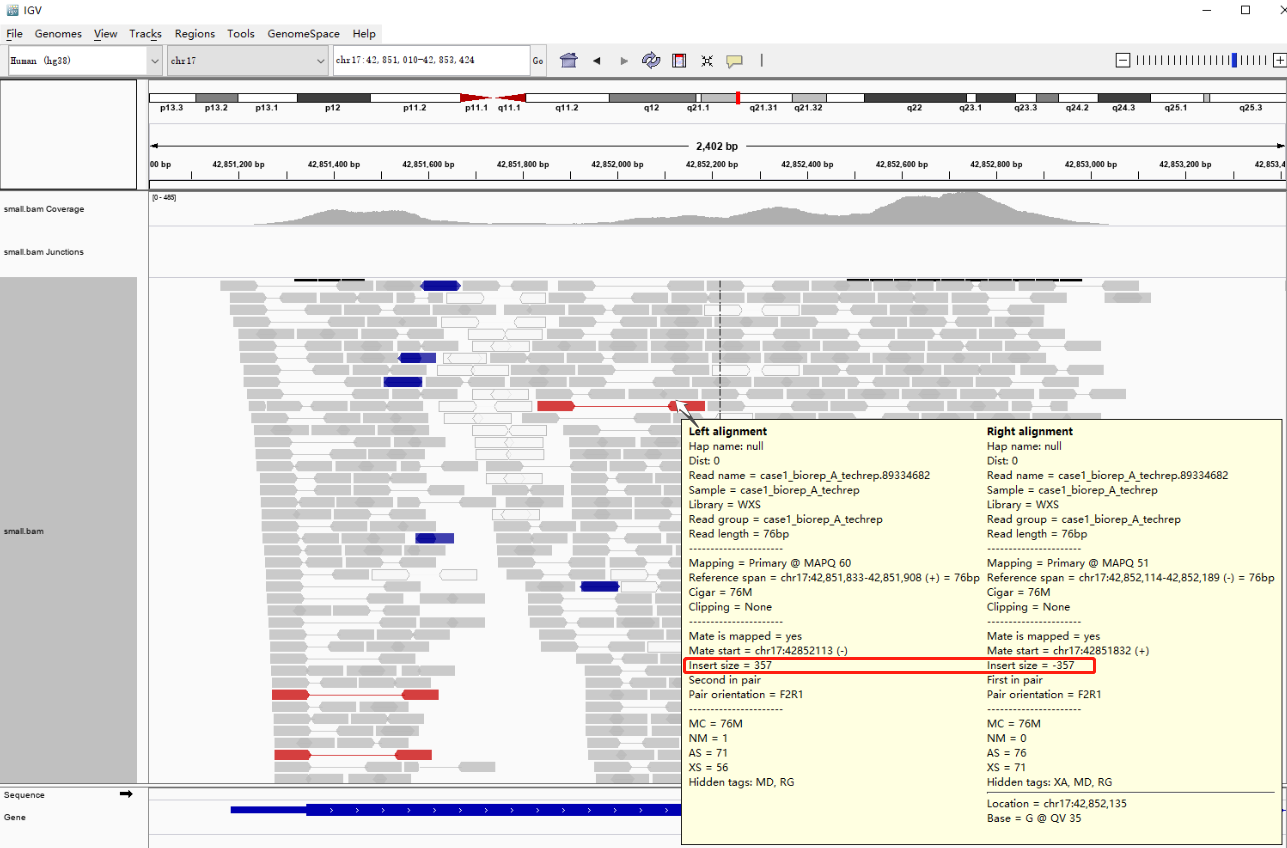
红色和蓝色: IGV 会根据配对的两条 reads 的距离,即 bam 的第 9 列 TLEN 测序是文库插入片段长度,来判断是否存在染色体的结构变异。红色代表插入片段大于期望值,可能是染色体片段缺失的证据,蓝色代表插入片段小于预期,可能是染色体插入的证据。我们可以通过右键选择 view as pairs 来进一步理解这个含义。蓝色的两条 reads 重叠,而红色的 reads 距离都比较大
其他颜色:指的是这条 reads 所配对的另一条 reads 没有比对到同一条染色体,不同颜色代表不同染色体,具体看下图:

比如下面棕色的 reads ,代表与之配对的reads比对到了 11 号染色体上了:
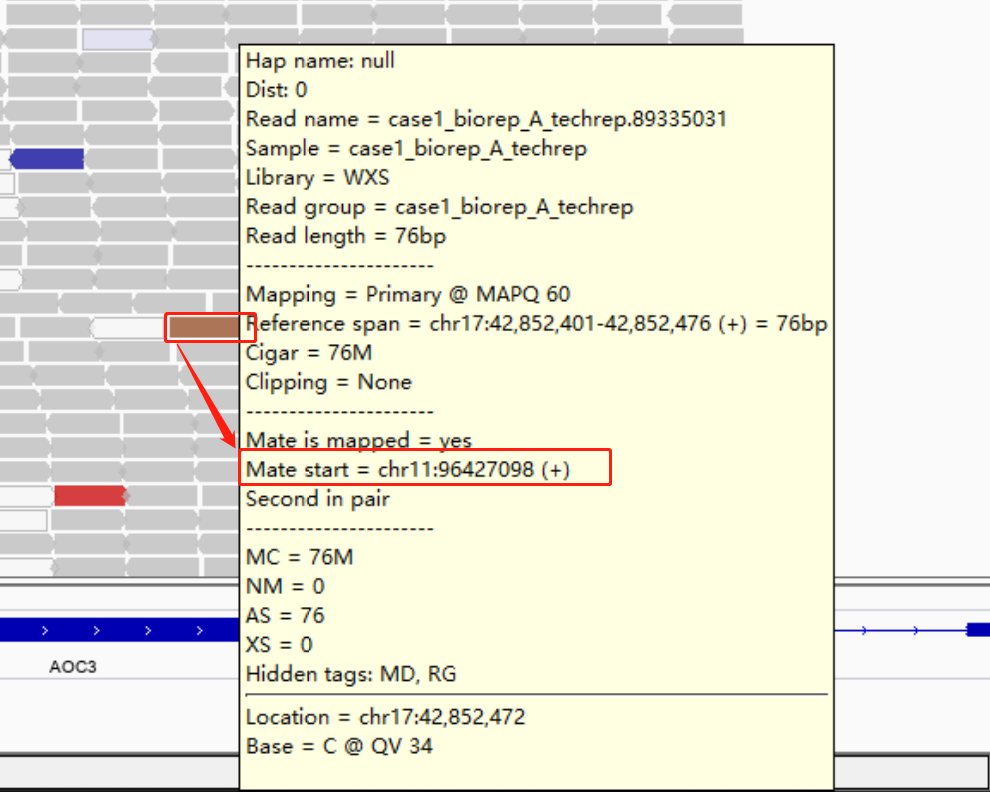
我们可以通过 bam 文件来检验一下,因为是 Pair-end,所以 id 号相同,可以 grep 89335031:
$ samtools view -h case1_biorep_A_techrep.bam chr11 | grep 89335031
case1_biorep_A_techrep.89335031 65 chr11 96427099 60 76M chr17 42852401 0 AAATTGAATCTGCAATTTCTCAACCCATTAAATTGTTCATCAATGCTGAACTAATACAAGAGTTACATTAATAAGC @>>??G?@AEDEDCAAAADCECADBBCAAABA?@FAAECAEBA@EEDCAADDABAADCBEAF@A@EC@@=A@>@DD NM:i:0 MD:Z:76 MC:Z:76M AS:i:76 XS:i:19 RG:Z:case1_biorep_A_techrep
$ samtools view -h case1_biorep_A_techrep.bam chr17 | grep 89335031
case1_biorep_A_techrep.89335031 129 chr17 42852401 60 76M chr11 96427099 0 GCTTCCAAGGAGAAAGACTAGTTTATGAGATAAGCCTCCAAGAGGCCTTGGCCATCTATGGTGGAAATTCCCCAGC @@@?DDCAFCAEABAE@DC@E@@@@ADAFAAAAEEBCDCBAFAFDECDAFDEDBAEDBBFD@GD@AAA@E@CA@DD NM:i:0 MD:Z:76 MC:Z:76M AS:i:76 XS:i:0 RG:Z:case1_biorep_A_techrep
关于颜色更多的介绍,请参考 IGV 官方文档:http://software.broadinstitute.org/software/igv/interpreting_insert_size
不同组学的测序策略不同,这里展示的是外显子组的 bam 文件,还有其他组学的 bam 文件也可以在 IGV 中可视化,对此推荐大家看一下不同组学测序数据拿到的bam文件在 IGV 可视化结果的区别:有参组学的NGS数据分析的异同点分享

若有收获,就点个赞吧
0 人点赞
