官网文档:https://support.sentieon.com/manual/_downloads/Sentieon.pdf
DNAseq基本流程
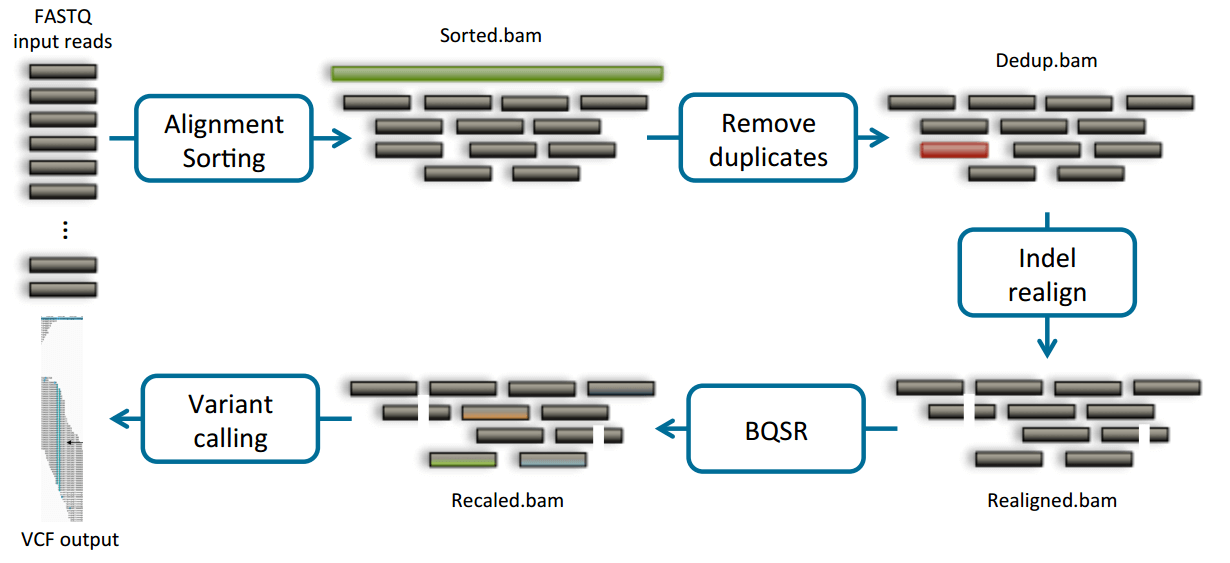
1 realign- Realigner
nodup.bam -> realn.bam
sentieon driver \-t NUMBER_THREADS \-i NODUP_BAM \\-r REFERENCE \--algo Realigner \-k MILLS_INDELS \REALIGNED_BAM
2 recal(BQSR) - QualCal
realn.bam -> recal.table [+ recal.bam]
# 仅生成recal.table [推荐]sentieon driver \-t NUMBER_THREADS \-i REALIGNED_BAM \-r REFERENCE \--algo QualCal \-k MILLS_INDELS \-k DBSNP \RECAL_DATA.TABLE# 同时生成recal.bamsentieon driver \-t NUMBER_THREADS \-i REALIGNED_BAM \-r REFERENCE \--algo QualCal \-k MILLS_INDELS \-k DBSNP \RECAL_DATA.TABLE \--algo ReadWriter \RECALIBRATED_BAM
3 Variant calling - Haplotyper / Genotyper
此步可分染色体进行,然后再用bcftools concat进行合并
方式1:使用Haplotyper算法,输入文件为 realn.bam + recal.table
sentieon driver \-t NUMBER_THREADS \-r REFERENCE \-i REALIGNED_BAM \-q RECAL_DATA.TABLE \--algo Haplotyper \--emit_mode gvcf \--emit_conf=10 \--call_conf=10 \[-d dbSNP] \VARIANT_GVCF
方式2: 使用Genotyper算法,输入文件为recal.bam
sentieon driver-t NUMBER_THREADS \-r REFERENCE \-i RECALIBRATED_BAM \--algo Genotyper \[-d dbSNP] \--emit_mode gvcf \VARIANT_GVCF
PS: 注意方式2中不能使用recal.bam+recal.table作为输入,否则会recal两次而导致错误结果
4 joint calling - GVCFtyper
s1.g.vcf.gz, s2.g.vcf.gz, … -> merged.vcf.gz
把一个或多个样本的GVCF合并成一个VCF文件, 输入文件可以通过-v指定
sentieon driver \-t NUMBER_THREADS \-r REFERENCE \--algo GVCFtyper \-d DBSNP \-v s1.g.vcf.gz \-v s2.g.vcf.gz \merge.vcf.gz
也可以直接在输出文件后面列上
sentieon driver -r reference.fasta [driver_options] \--algo GVCFtyper [algo_options] output.vcf[.gz] input.g.vcf[.gz] ...
5 VQSR - VarCal + ApplyVarCal
VQSR: Variant Quality Score Recalibration 使用一些可信的已知位点构建模型,对检出的变异位点进行过滤 merge.vcf.gz -> SNP VQSR -> Apply SNP VQSR -> INDEL VQSR -> Apply INDEL VQSR -> out.vcf.gz
# Step1: SNP VQSRsentieon driver \\-t {vqsr_threads} \\-r {reffasta} \\--algo VarCal \\--var_type SNP \\--resource {1000g_phase1} \\--resource_param 1000G,known=false,training=true,truth=false,prior=10.0 \\--resource {1000g_omni} \\--resource_param omni,known=false,training=true,truth=true,prior=12.0 \\--resource {dbsnp} \\--resource_param dbsnp,known=true,training=false,truth=false,prior=2.0 \\--resource {hapmap} \\--resource_param hapmap,known=false,training=true,truth=true,prior=15.0 \\--annotation QD --annotation MQ --annotation MQRankSum \\--annotation ReadPosRankSum --annotation FS \\--tranches_file VCF/all.merged.snp.tranches \\-v VCF/all.merged.vcf.gz \\VCF/all.merged.snp.recal# Step2: Apply SNP VQSRsentieon driver \\-t {vqsr_threads} \\-r {reffasta} \\--algo ApplyVarCal \\--var_type SNP \\--recal VCF/all.merged.snp.recal \\--tranches_file VCF/all.merged.snp.tranches \\-v VCF/all.merged.vcf.gz \\VCF/all.merged.snp.recal.vcf.gz# Step3: INDEL VQSRsentieon driver \\-t {vqsr_threads} \\-r {reffasta} \\--algo VarCal \\--var_type INDEL \\--resource {dbsnp} \\--resource_param dbsnp,known=true,training=false,truth=false,prior=2.0 \\--resource {mills_indels} \\--resource_param Mills,known=false,training=true,truth=true,prior=12.0 \\--annotation QD --annotation MQ \\--annotation ReadPosRankSum --annotation FS \\--tranches_file VCF/all.merged.snp.indel.tranches \\-v VCF/all.merged.snp.recal.vcf.gz \\VCF/all.merged.snp.indel.recal# Step4: Apply INDEL VQSRsentieon driver \\-t {vqsr_threads} \\-r {reffasta} \\--algo ApplyVarCal \\--var_type INDEL \\--recal VCF/all.merged.snp.indel.recal \\--tranches_file VCF/all.merged.snp.indel.tranches \\-v VCF/all.merged.snp.recal.vcf.gz \\VCF/all.merged.snp.indel.recal.vcf.gz

若有收获,就点个赞吧
0 人点赞
