1 基因组
1.1 fasta基因组下载
http://hgdownload.cse.ucsc.edu/goldenPath/rn6/bigZips/
下载rn6.fa.gz即可(repeats标记为小写,而没有标记为N)
1.2 统计文件
每条染色体长度和含N数量统计,可通过脚本genome_stat计算
genome_stat rn6.fa# 生成 rn6.fa.bed 和 rn6.fa.nblocks# 用于深度统计等
其他获取每条染色体长度的方法:
- 下载rn6.chrom.sizes文件,自行转成bed格式
- 从rn6.fa.fai文件(参见下方索引文件)中获取,再转成bed
1.3 索引文件
```bashfor samtools
samtools faidx rn6.fa
for bwa
bwa index -a bwtsw rn6.fa
for crest
faToTwoBit rn6.fa rn6.fa.2bit
.2bit文件也可以直接下载[rn6.2bit](http://hgdownload.cse.ucsc.edu/goldenPath/rn6/bigZips/rn6.2bit)<br />`faToTwoBit`为[**ucsc工具包**](http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/)中的一个命令[**Control-FREEC**](http://boevalab.com/FREEC/tutorial.html)需要的文件- rn6.fa.length - 每条染色体的长度 `cut -f 1,3 rn6.fa.bed | grep -v M > rn6.fa.length`- bychr - 包含每条染色体fa序列的目录- gemMappabilityFile - WGS without Control时需要的gem文件- rn6.fa.nblocks.gte1000bp.bed - 染色体中长度≥1000的Nblocks区间([get_Nblock.py]())```bashcut -f 1,3 rn6.fa.bed | grep -v M > rn6.fa.length # 只保留1-Y# 按染色体拆分fa文件mkdir -p bychrfor chr in chr{{1..20},X,Y,M};doecho samtools-1.9 faidx rn6.fa $chr -o bychr/${chr}.fadone > jobs2runparallel -j4 < jobs2run# 获取N的bed区间python get_Nblock.py rn6.fa > rn6.fa.nblocks.bedawk '($3-$2)>=1000' rn6.fa.nblocks.bed > rn6.fa.nblocks.gte1000bp.bed
通过GEM创建基因组Mappability文件
gem-indexer -T 8 -c dna -i ../rn6.fa -o rn6# 生成 rn6.gemgem-mappability -T 8 -I rn6.gem -l 150 -o rn6.150bp# 生成 rn6.150bp.mappability
2 Annovar注释文件
http://hgdownload.cse.ucsc.edu/goldenPath/rn6/database/ 如refGene, cytoBand, cpgIslandExt, rmsk等可用于构建annovar注释库文件
index_annovar.pl不是annovar的官方脚本,这里有个可用的脚本备份(不知道从哪里下载的)
2.1 Repeat
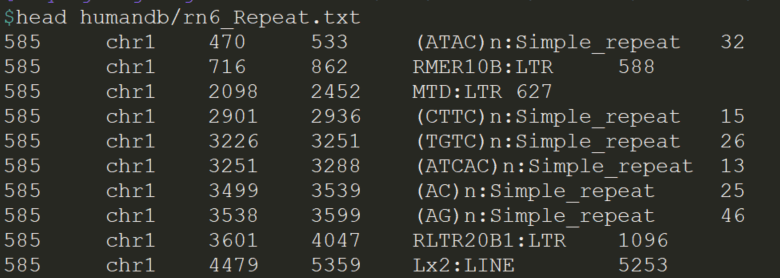
rmsk.txt.gz 重复序列信息注释, 重复序列来源于RepeatMasker注释
mkdir -p humandbzcat rmsk.txt.gz | awk -F '\t' '{printf "%s\t%s\t%s\t%s\t%s:%s\t%s\n",$1,$6,$7,$8,$11,$12,$2}' > rn6_Repeat.txtindex_annovar.pl \--filetype B \--outfile humandb/rn6_Repeat.txt \rn6_Repeat.txtrm -f rn6_Repeat.txt# annovar 注释参数arguments: --scorecolumn 5 # 第6列注释为Score=xxx, 第5列注释为Name=xxx
2.2 refGene
refGene.txt.gz 基于gene的注释,会注释以下5列 Func 变异位点所在的区域 Gene 变异位点相关的转录本 GeneDetail 描述UTR、splicing、ncRNA_splicing或intergenic区域的变异情况 ExonicFunc 外显子区的SNV 或 InDel变异类型 AAChange 氨基酸改变
refGene的注释需要有对应的refGeneMrna.fa序列文件,如果没有可通过retrieve_seq_from_fasta.pl命令自行生成
pigz -kd refGene.txt.gzindex_annovar.pl \--filetype C \--outfile humandb/rn6_refGene.txt \refGene.txtrm -f refGene.txtretrieve_seq_from_fasta.pl \--format refGene \--seqfile /path/to/rn6.fa \--outfile humandb/rn6_refGeneMrna.fa \
refGene每列信息说明(http://genome.ucsc.edu/cgi-bin/hgTables)
| field | example | SQL type | info | description | |
|---|---|---|---|---|---|
| 1 | bin | 585 | smallint(5) unsigned | range | Indexing field to speed chromosome range queries. |
| 2 | name | XR_001835498.1 | varchar(255) | values | Name of gene (usually transcript_id from GTF) |
| 3 | chrom | chr1 | varchar(255) | values | Reference sequence chromosome or scaffold |
| 4 | strand | - | char(1) | values | + or - for strand |
| 5 | txStart | 53395 | int(10) unsigned | range | Transcription start position (or end position for minus strand item) |
| 6 | txEnd | 56311 | int(10) unsigned | range | Transcription end position (or start position for minus strand item) |
| 7 | cdsStart | 56311 | int(10) unsigned | range | Coding region start (or end position for minus strand item) |
| 8 | cdsEnd | 56311 | int(10) unsigned | range | Coding region end (or start position for minus strand item) |
| 9 | exonCount | 3 | int(10) unsigned | range | Number of exons |
| 10 | exonStarts | 53395,55034,55623, | longblob | Exon start positions (or end positions for minus strand item) | |
| 11 | exonEnds | 53701,55222,56311, | longblob | Exon end positions (or start positions for minus strand item) | |
| 12 | score | 0 | int(11) | range | score |
| 13 | name2 | LOC108349478 | varchar(255) | values | Alternate name (e.g. gene_id from GTF) |
| 14 | cdsStartStat | none | enum(‘none’, ‘unk’, ‘incmpl’, ‘cmpl’) | values | enum(‘none’,’unk’,’incmpl’,’cmpl’) |
| 15 | cdsEndStat | none | enum(‘none’, ‘unk’, ‘incmpl’, ‘cmpl’) | values | enum(‘none’,’unk’,’incmpl’,’cmpl’) |
| 16 | exonFrames | -1,-1,-1, | longblob | Exon frame {0,1,2}, or -1 if no frame for exon |
2.3 GeneName
只是从refGene里把基因名(name2)拆出来了,即NCBI的标准名
les refGene.txt.gz |awk -v OFS='\t' -F '\t' '{print $1,$3,$5,$6,0,$13}' > GeneName.txtindex_annovar.pl --filetype B --outfile humandb/rn6_GeneName.txt GeneName.txtrm -f GeneName.txt
2.4 Gencode
Gencode只有人和鼠的,用Ensembl代替:ftp://ftp.ensembl.org/pub/current_gff3 下载Rattus_norvegicus.Rnor_6.0.98.gff3.gz文件,提取区间和ID
ens2annvoar Rattus_norvegicus.Rnor_6.0.98.gff3.gz -o rn6_Gencode.txt
2.5 dbsnp
NCBI Taxonomy ID查询:https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi
从NCBI下载 00-All.vcf.gz
rat_10116 - Rattus Norvegicus rat_10118 - Rattus Sp
ftp://ftp.ncbi.nih.gov/snp/organisms/archive/rat_10116/
https://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?do_not_redirect&rs=rs105383212
convert2annovar.pl -format vcf4 -includeinfo 00-All.vcf.gz |awk -v OFS='\t' '{print $1,$2,$3,$4,$5,$8}' > dbsnp149.txtindex_annovar.pl \--filetype A \--outfile humandb/rn6_dbsnp149.txt \dbsnp149.txtrm -f dbsnp149.txt
2.6 cytoBand,cpgIslandExt
可直接使用的 cytoBand.txt.gz 变异位点所处的染色体区段 cpgIslandExt.txt.gz CpG岛预测结果
pigz -kd cytoBand.txt.gzindex_annovar.pl \--filetype A \--outfile humandb/rn6_cytoBand.txt \cytoBand.txtrm -f cytoBand.txtpigz -kd cpgIslandExt.txt.gzindex_annovar.pl \--filetype B \--outfile humandb/rn6_cpgIslandExt.txt \cpgIslandExt.txtrm -f cpgIslandExt.txt

若有收获,就点个赞吧
0 人点赞
