- Single cell RNA-seq Data processing
- Downlaod Practice Data
- Data loading
- Lets examine a few genes in the first thirty cells
- Standard pre-processing workflow
- Data Normalization
- Identify of highly variable features (feature selection)
- Scaling the data
- Perform linear dimensional reduction
- Determine the ‘dimensionality’ of the dataset
- Cluster the cells
- Run non-linear dimensional reduction (UMAP/tSNE)
- Finding differentially expressed features (cluster biomarkers)
- Assigning cell type identity to clusters
| © Karobben |
由於語法渲染問題而影響閱讀體驗, 請移步博客閱讀~
本文GitPage地址
Single cell RNA-seq Data processing
This is my first time to learn siRNA-Seq. The protocol are based on Seurat. Please go and reading more information from Seurat. The codes are derectly copied from Seurat and so, if you are confuzed about my moves, please go to the link below and check by yourselves.
For new users of Seurat, we suggest starting with a guided walk through of a dataset of 2,700 Peripheral Blood Mononuclear Cells (PBMCs) made publicly available by 10X Genomics. This tutorial implements the major components of a standard unsupervised clustering workflow including QC and data filtration, calculation of high-variance genes, dimensional reduction, graph-based clustering, and the identification of cluster markers.
—Seurat
Source code: © Seurat 2021
Downlaod Practice Data
Data set: Peripheral Blood Mononuclear Cells (PBMC) freely available from 10X Genomics.
wget https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gztar -zxvf pbmc3k_filtered_gene_bc_matrices.tar.gztree filtered_gene_bc_matrices/hg19
filtered_gene_bc_matrices/hg19├── barcodes.tsv├── genes.tsv└── matrix.mtx
head -n 4 filtered_gene_bc_matrices/hg19/*
==> filtered_gene_bc_matrices/hg19/barcodes.tsv <==
AAACATACAACCAC-1
AAACATTGAGCTAC-1
AAACATTGATCAGC-1
AAACCGTGCTTCCG-1
==> filtered_gene_bc_matrices/hg19/genes.tsv <==
ENSG00000243485 MIR1302-10
ENSG00000237613 FAM138A
ENSG00000186092 OR4F5
ENSG00000238009 RP11-34P13.7
==> filtered_gene_bc_matrices/hg19/matrix.mtx <==
%%MatrixMarket matrix coordinate real general
%
32738 2700 2286884
32709 1 4
Data loading
library(dplyr)
library(Seurat)
library(patchwork)
# Load the PBMC dataset
pbmc.data <- Read10X(data.dir = "filtered_gene_bc_matrices/hg19/")
# Initialize the Seurat object with the raw (non-normalized data).
pbmc <- CreateSeuratObject(counts = pbmc.data, project = "pbmc3k", min.cells = 3, min.features = 200)
pbmc
An object of class Seurat
13714 features across 2700 samples within 1 assay
Active assay: RNA (13714 features, 0 variable features)
What does data in a count matrix look like?```r
Lets examine a few genes in the first thirty cells
pbmc.data[c(“CD3D”, “TCL1A”, “MS4A1”), 1:30]
```
3 x 30 sparse Matrix of class "dgCMatrix"
CD3D 4 . 10 . . 1 2 3 1 . . 2 7 1 . . 1 3 . 2 3 . . . . . 3 4 1 5
TCL1A . . . . . . . . 1 . . . . . . . . . . . . 1 . . . . . . . .
MS4A1 . 6 . . . . . . 1 1 1 . . . . . . . . . 36 1 2 . . 2 . . . .
dense.size <- object.size(as.matrix(pbmc.data))
dense.size
sparse.size <- object.size(pbmc.data)
sparse.size
dense.size/sparse.size
709591472 bytes
29905192 bytes
23.7 bytes
Standard pre-processing workflow
QC and selecting cells for further analysis
# The [[ operator can add columns to object metadata. This is a great place to stash QC stats
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
Where are QC metrics stored in Seurat?```r head(pbmc@meta.data, 5)
```
<table class="dataframe">
<thead>
<tr><th></th><th scope="col">orig.ident</th><th scope="col">nCount_RNA</th><th scope="col">nFeature_RNA</th><th scope="col">percent.mt</th></tr>
<tr><th></th><th scope="col"><fct></th><th scope="col"><dbl></th><th scope="col"><int></th><th scope="col"><dbl></th></tr>
</thead>
<tbody>
<tr><th scope="row">AAACATACAACCAC-1</th><td>pbmc3k</td><td>2419</td><td> 779</td><td>3.0177759</td></tr>
<tr><th scope="row">AAACATTGAGCTAC-1</th><td>pbmc3k</td><td>4903</td><td>1352</td><td>3.7935958</td></tr>
<tr><th scope="row">AAACATTGATCAGC-1</th><td>pbmc3k</td><td>3147</td><td>1129</td><td>0.8897363</td></tr>
<tr><th scope="row">AAACCGTGCTTCCG-1</th><td>pbmc3k</td><td>2639</td><td> 960</td><td>1.7430845</td></tr>
<tr><th scope="row">AAACCGTGTATGCG-1</th><td>pbmc3k</td><td> 980</td><td> 521</td><td>1.2244898</td></tr>
</tbody>
</table>
 |
|---|
| ©Seurat 2021 |
At here, we filter cells that have:
- unique feature counts over 2,500 or less than 200
5% mitochondrial counts
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
 |
|---|
| ©Seurat 2021 |
dim(pbmc)
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
dim(pbmc)
[1] 13714 2700
[1] 13714 2638
Data Normalization
By default, we employ a global-scaling normalization method “LogNormalize” that normalizes the feature expression measurements for each cell by the total expression, multiplies this by a scale factor (10,000 by default), and log-transforms the result. Normalized values are stored in pbmc[[“RNA”]]@data.
# pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)
pbmc <- NormalizeData(pbmc)
Performing log-normalization
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
Identify of highly variable features (feature selection)
Seurat believe that “focusing on these genes in downstream analysis helps to highlight biological signal in single-cell datasets”
Identification argorithm details: Comprehensive Integration of Single-Cell Data
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)
# Identify the 10 most highly variable genes
top10 <- head(VariableFeatures(pbmc), 10)
# plot variable features with and without labels
plot1 <- VariableFeaturePlot(pbmc)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
plot1 + plot2
Scaling the data
Next, we apply a linear transformation (‘scaling’) that is a standard pre-processing step prior to dimensional reduction techniques like PCA. The ScaleData() function:
Shifts the expression of each gene, so that the mean expression across cells is 0
Scales the expression of each gene, so that the variance across cells is 1
This step gives equal weight in downstream analyses, so that highly-expressed genes do not dominate
The results of this are stored in pbmc[["RNA"]]@scale.data
all.genes <- rownames(pbmc)
pbmc <- ScaleData(pbmc, features = all.genes)
Perform linear dimensional reduction
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc)
# Examine and visualize PCA results a few different ways
print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")
 |
|---|
| ©Seurat 2021 |
DimPlot(pbmc, reduction = "pca")
 |
|---|
| ©Seurat 2021 |
DimHeatmap(pbmc, dims = 1:15, cells = 500, balanced = TRUE)
 |
|---|
| ©Seurat 2021 |
Determine the ‘dimensionality’ of the dataset
# NOTE: This process can take a long time for big datasets, comment out for expediency. More
# approximate techniques such as those implemented in ElbowPlot() can be used to reduce
# computation time
pbmc <- JackStraw(pbmc, num.replicate = 100)
pbmc <- ScoreJackStraw(pbmc, dims = 1:20)
JackStrawPlot(pbmc, dims = 1:15)
 |
|---|
| ©Seurat 2021 |
ElbowPlot(pbmc)
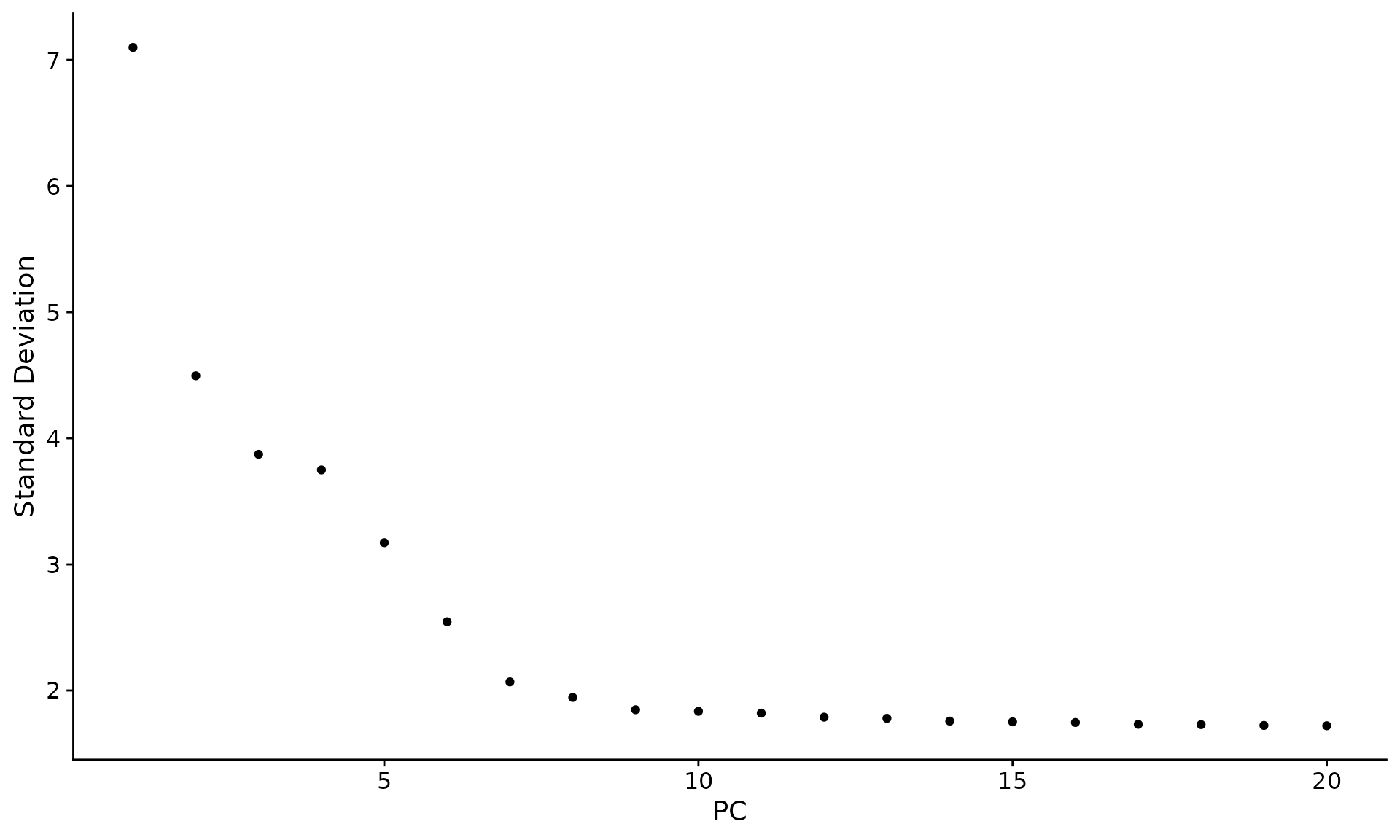 |
|---|
| ©Seurat 2021 |
Cluster the cells
pbmc <- FindNeighbors(pbmc, dims = 1:10)
pbmc <- FindClusters(pbmc, resolution = 0.5)
# Look at cluster IDs of the first 5 cells
head(Idents(pbmc), 5)
Run non-linear dimensional reduction (UMAP/tSNE)
# If you haven't installed UMAP, you can do so via reticulate::py_install(packages =
# 'umap-learn')
pbmc <- RunUMAP(pbmc, dims = 1:10)
DimPlot(pbmc, reduction = "umap")
saveRDS(pbmc, file = "../output/pbmc_tutorial.rds")
 |
|---|
| ©Seurat 2021 |
Finding differentially expressed features (cluster biomarkers)
cluster2.markers <- FindMarkers(pbmc, ident.1 = 2, min.pct = 0.25)
head(cluster2.markers, n = 5)
# find all markers distinguishing cluster 5 from clusters 0 and 3
cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25)
head(cluster5.markers, n = 5)
pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
pbmc.markers %>%
group_by(cluster) %>%
slice_max(n = 2, order_by = avg_log2FC)
cluster0.markers <- FindMarkers(pbmc, ident.1 = 0, logfc.threshold = 0.25, test.use = "roc", only.pos = TRUE)
VlnPlot(pbmc, features = c("MS4A1", "CD79A"))
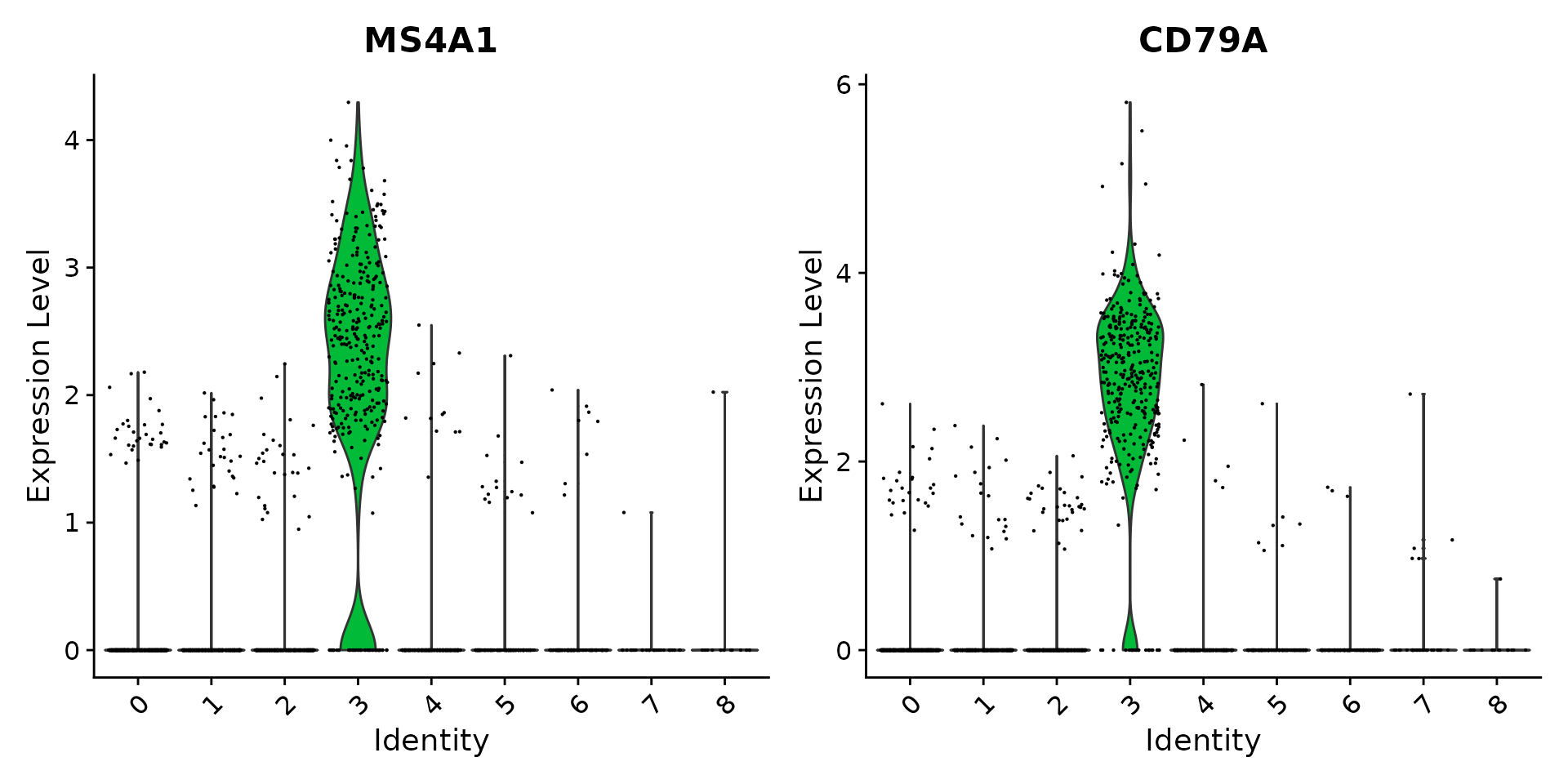 |
|---|
| ©Seurat 2021 |
# you can plot raw counts as well
VlnPlot(pbmc, features = c("NKG7", "PF4"), slot = "counts", log = TRUE)
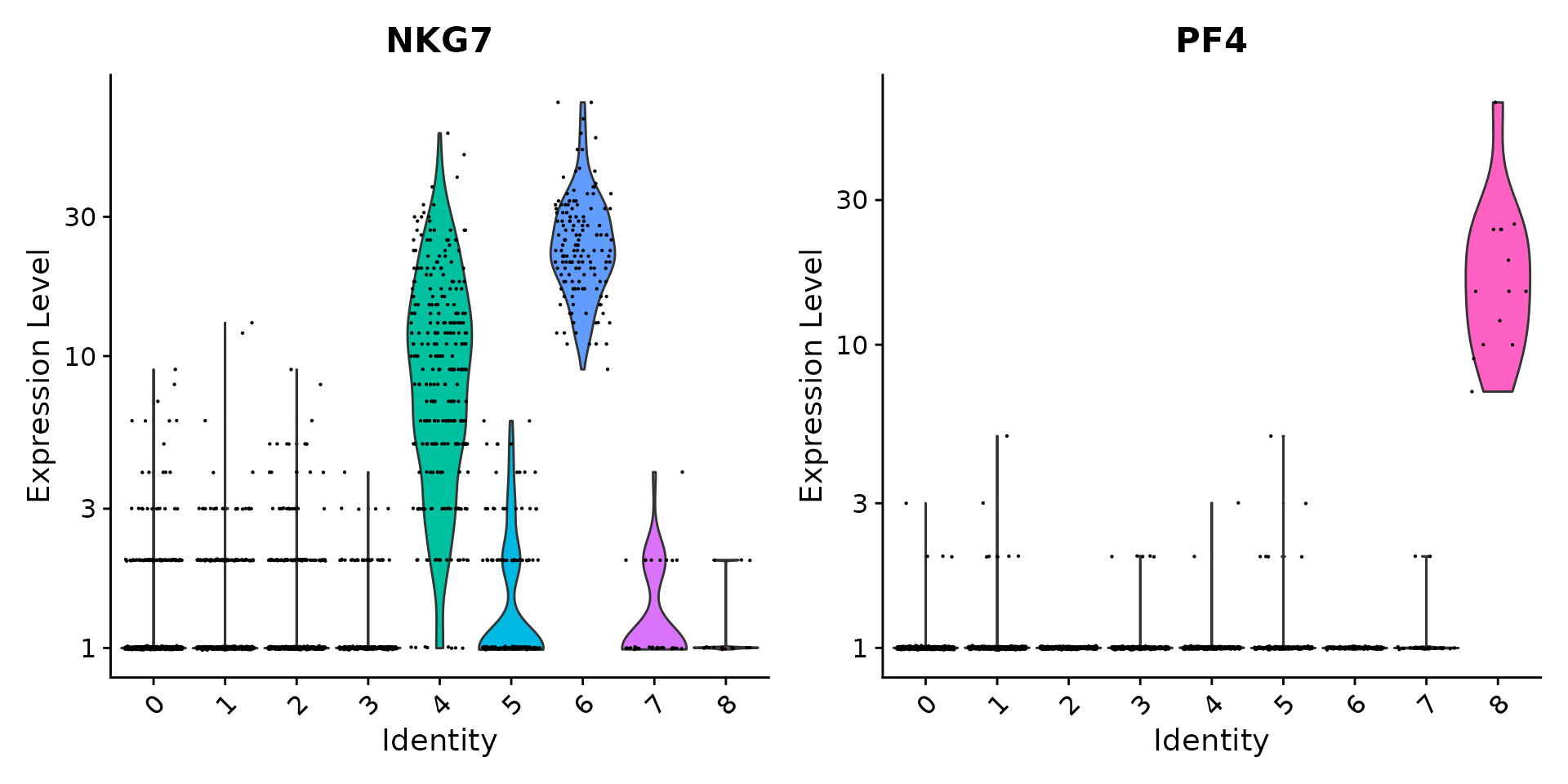 |
|---|
| ©Seurat 2021 |
FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP",
"CD8A"))
 |
|---|
| ©Seurat 2021 |
pbmc.markers %>%
group_by(cluster) %>%
top_n(n = 10, wt = avg_log2FC) -> top10
DoHeatmap(pbmc, features = top10$gene) + NoLegend()
 |
|---|
| ©Seurat 2021 |
Assigning cell type identity to clusters
new.cluster.ids <- c("Naive CD4 T", "CD14+ Mono", "Memory CD4 T", "B", "CD8 T", "FCGR3A+ Mono",
"NK", "DC", "Platelet")
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
saveRDS(pbmc, file = "../output/pbmc3k_final.rds")
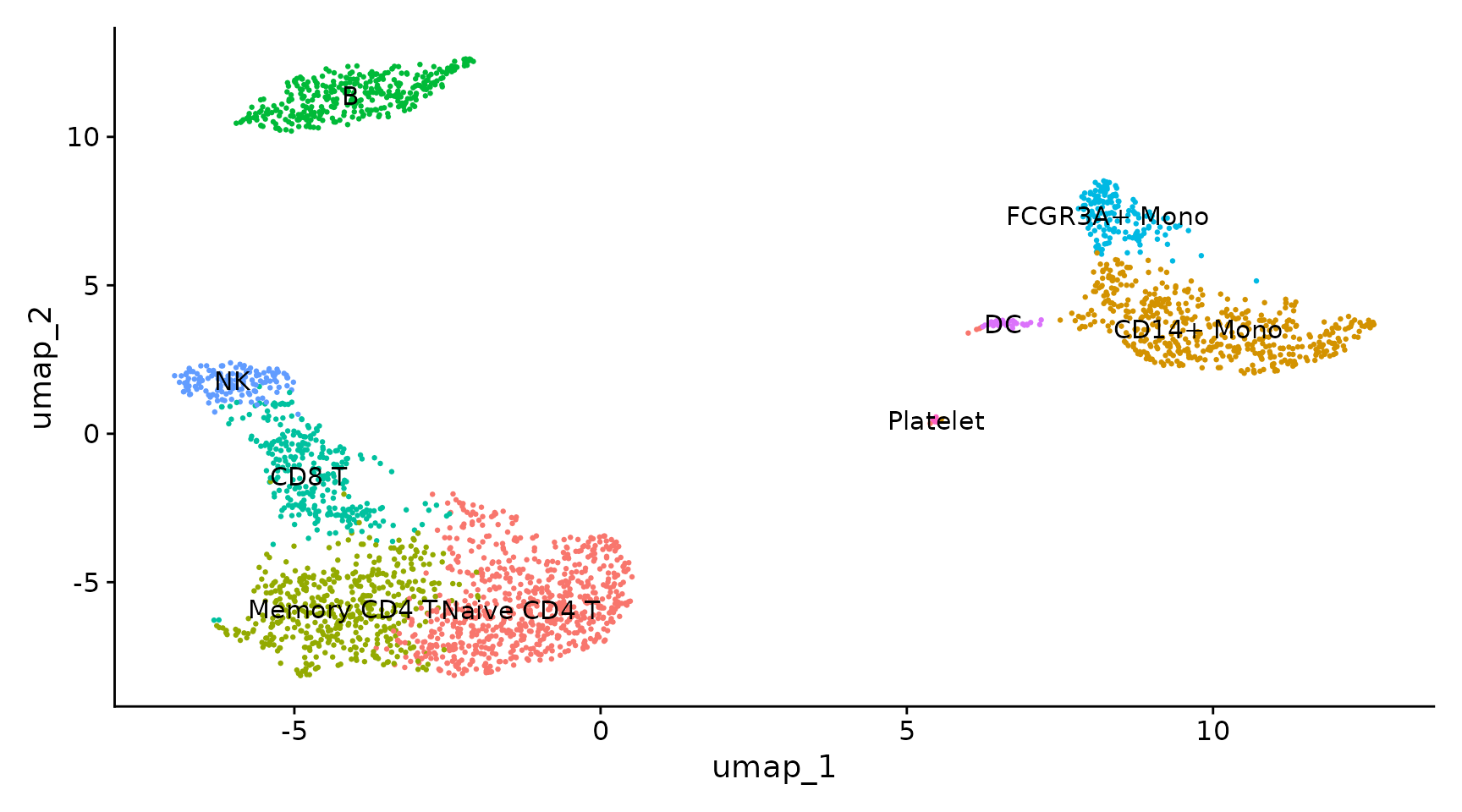 |
|---|
| ©Seurat 2021 |
Enjoy~
由於語法渲染問題而影響閱讀體驗, 請移步博客閱讀~
本文GitPage地址
GitHub: Karobben
Blog:Karobben
BiliBili:史上最不正經的生物狗

若有收获,就点个赞吧
0 人点赞
