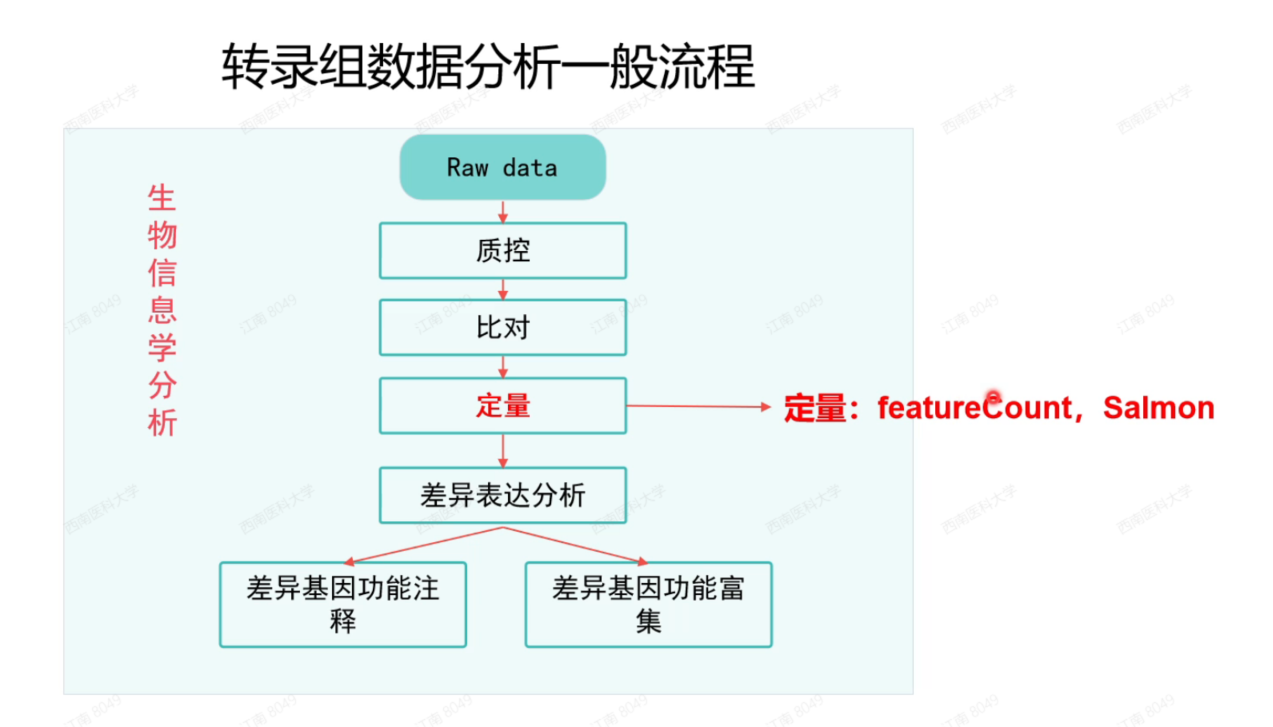
软件
featureCounts

官网:http://bioinf.wehi.edu.au/featureCounts/

cd $HOME/project/Human-16-Asthma-Trans/Expression/featureCounts## 定义输入输出文件夹gtf=/home/t_rna/database/GRCh38.104/Homo_sapiens.GRCh38.104.chr.gtf.gzinputdir=$HOME/project/Human-16-Asthma-Trans/Mapping/Hisat2/# featureCounts对bam文件进行计数featureCounts -T 6 -p -t exon -g gene_id -a $gtf -o all.id.txt $inputdir/*.sorted.bam# 对定量结果质控multiqc all.id.txt.summary# 得到表达矩阵# 处理表头,/home/t_rna/要换成自己的路径less -S all.id.txt |grep -v '#' |cut -f 1,7- |sed 's#/home/t_rna/project/Human-16-Asthma-Trans/Mapping/Hisat2//##g' |sed 's#.Hisat_aln.sorted.bam##g' >raw_counts.txtsed ///sed #### 列对齐显示head raw_counts.txt |column -t
salmon


直接定量
##----构建索引
## 后续索引可直接使用服务器上已经构建好的进行练习
cd $HOME/database/GRCh38.105
nohup salmon index -t Homo_sapiens.GRCh38.cdna.all.fa -i salmon_index >salmon-index.log &
cd $HOME/project/Human-16-Asthma-Trans/Expression/Salmon
##----运行
# 编写脚本,使用salmon批量对目录下所有fastq文件进行定量
# vim salmon.sh
index=/home/t_rna/database/GRCh38.104/salmon_index
input=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
outdir=$HOME/project/Human-16-Asthma-Trans/Expression/Salmon
cat ../../data/cleandata/trim_galore/ID |while read id
do
salmon quant -i ${index} -l A -1 ${input}/${id}_1_val_1.fq.gz -2 ${input}/${id}_2_val_2.fq.gz -p 5 -o ${outdir}/${id}.quant
done
##----合并表达矩阵
# 原始count值矩阵
# --quants:ls -d *quant |tr '\n' ',' |sed 's/,$//' |awk '{print "{"$0"}"}'
# --quants:ls -d *quant |sed ':a;N;s/\n/,/;t a;'|awk '{print "{"$0"}"}'
# --quants:ls -d *quant |xargs |tr ' ' ',' |awk '{print "{"$0"}"}'
# --names:ls -d *quant |tr '\n' ',' |sed 's/,$//' |awk '{print "{"$0"}"}' |sed 's/.quant//g'
## 完整版:ls -d *quant |tr '\n' ',' |sed 's/,$//' |awk '{print "{"$0"}"}' |perl -ne 'chomp;$a=$_;$a=~s/\.quant//g;print"salmon quantmerge --quants $_ --names $a --column numreads --output raw_count.txt \n";'
salmon quantmerge --quants {SRR1039510.quant,SRR1039511.quant,SRR1039512.quant} --names {SRR1039510,SRR1039511,SRR1039512} --column numreads --output raw_count.txt
# tpm矩阵
salmon quantmerge --quants {SRR1039510.quant,SRR1039511.quant,SRR1039512.quant} --names {SRR1039510,SRR1039511,SRR1039512} --column tpm --output tpm.txt
# 后台运行脚本
nohup sh salmon.sh >salmon.log &
总结:
1 raw data : fq文件 Q20 Q30, GC%, reads, bases
2, 质控 tramglore , fastq
3, fa gtf (ID — name) hisat2, salmon
4, faturecounts, salmon

若有收获,就点个赞吧
0 人点赞
