标准流程:
参考:https://satijalab.org/seurat/v3.0/pbmc3k_tutorial.html
数据:https://s3-us-west-2.amazonaws.com/10x.files/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
1 加载R包
rm(list = ls())options(stringsAsFactors = F)library(dplyr)library(Seurat)library(patchwork)
2 读取数据-》 创建seurat对象-》查看表达矩阵
10X的输入数据是固定的三个文件
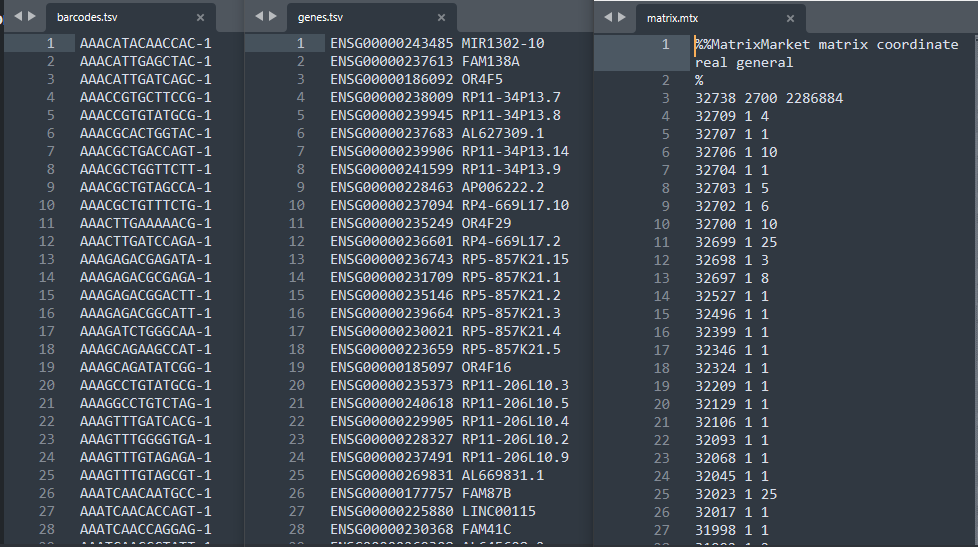
- barcodes.tsv是对细胞的编号,
- genes.tsv是测序基因注释
- matrix.mtx是表达数据
在工作目录下新建01_data/,把三个文件放进去。用read10X()读入成pbmc.data 一个矩阵 是dgcmatrix类
pbmc.data <- Read10X(data.dir = "01_data/")
dim(pbmc.data)
class(pbmc.data)
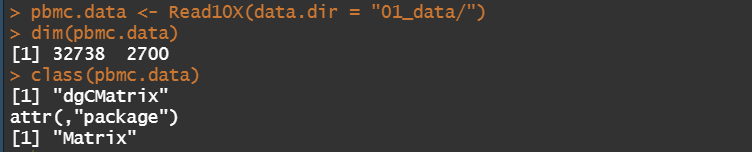
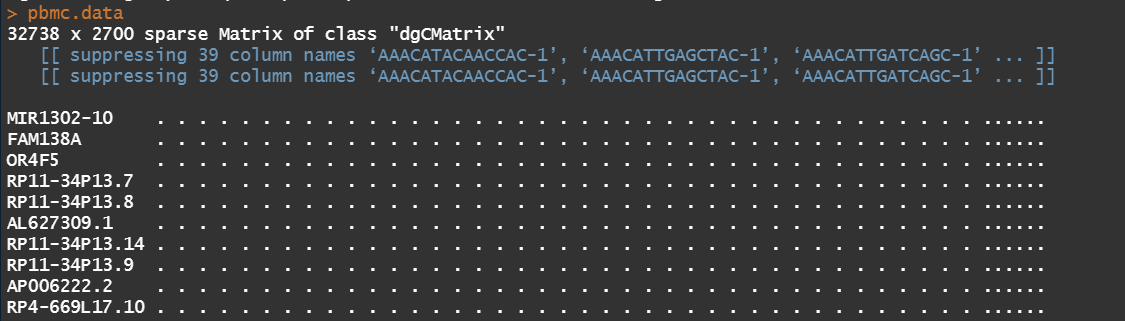
pdmc.data是一个稀疏矩阵 32738行(基因)* 2700列(细胞)
创建seurat对象,CreateSeuratObject ();参数:counts 是表达矩阵(一般分析从这里开始) 和project =“起一个名字”,min.cells 基因(行)小于多少个细胞中表达就不要了,min.features 某细胞(列)小于多少个基因就不要的
pbmc <- CreateSeuratObject(counts = pbmc.data,
project = "pbmc3k",
min.cells = 3,
min.features = 200)
pbmc

32738—》13714 表达量少的基因就不要了,所有2700个细胞都保留
查看表达矩阵
exp = pbmc@assays$RNA@counts;dim(exp)
exp[30:34,1:4]
boxplot(as.matrix(exp[,1:20]))
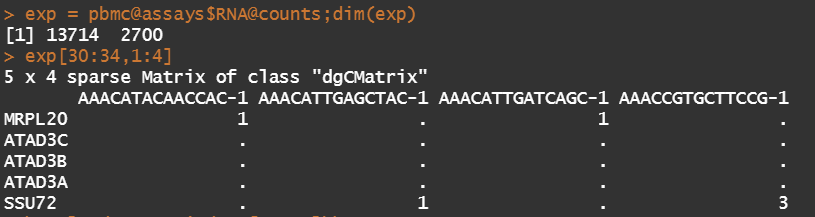
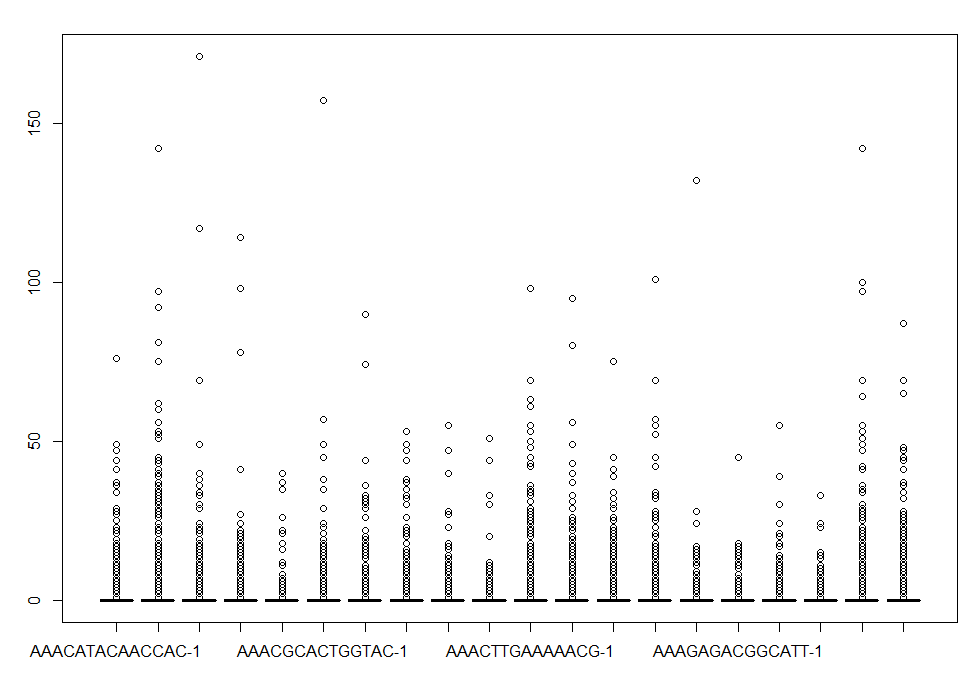
点号为0,该矩阵为稀疏矩阵,boxplot查看前20列表达,绝大多数基因表达为零,毕竟是单个细胞的表达
3 质控 (除去大多数双峰/死细胞/空液滴)
标准:
- 线粒体基因含量不能过高;
- nFeature_RNA 不能过高或过低,
nFeature_RNA是每个细胞中检测到的基因数量。nCount_RNA是细胞内检测到的分子总数。nFeature_RNA过低,表示该细胞可能已死/将死或是空液滴。太高的nCount_RNA和/或nFeature_RNA表明“细胞”实际上可以是两个或多个细胞。结合线粒体基因count数除去异常值,即可除去大多数双峰/死细胞/空液滴,因此它们过滤是常见的预处理步骤。 参考自:https://www.biostars.org/p/407036/
PercentageFeatureSet()在pbmc里面正则表达计算MT-开头的基因比例 并 增加 percent.mt属性
VlnPlot ()seurat 自带作图函数 统计可视化每个细胞 3个feature,可按样本分组显示,
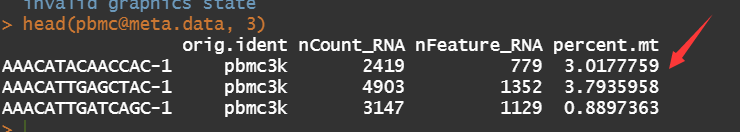
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
head(pbmc@meta.data, 3)
VlnPlot(pbmc,
features = c("nFeature_RNA",
"nCount_RNA",
"percent.mt"),
ncol = 3, pt.size=0.1)
# 过滤
pbmc <- subset(pbmc,
subset = nFeature_RNA > 200 &
nFeature_RNA < 2500 &
percent.mt < 5)
dim(pbmc)
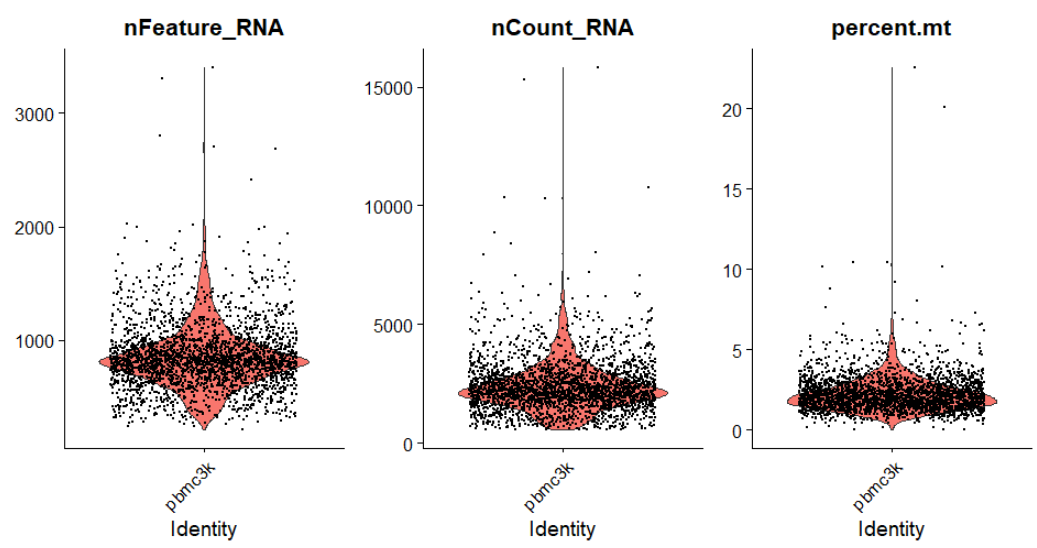
根据这个三个图,确定了这个数据的细胞过滤标准:
- nFeature_RNA在200~2500之间;
- 线粒体基因占比在5%以下。
- 过滤用subset(),参数subset= XX & xx
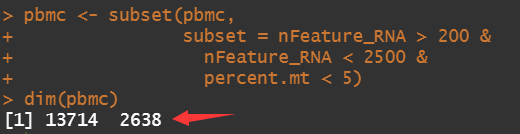 过滤掉62个细胞
过滤掉62个细胞
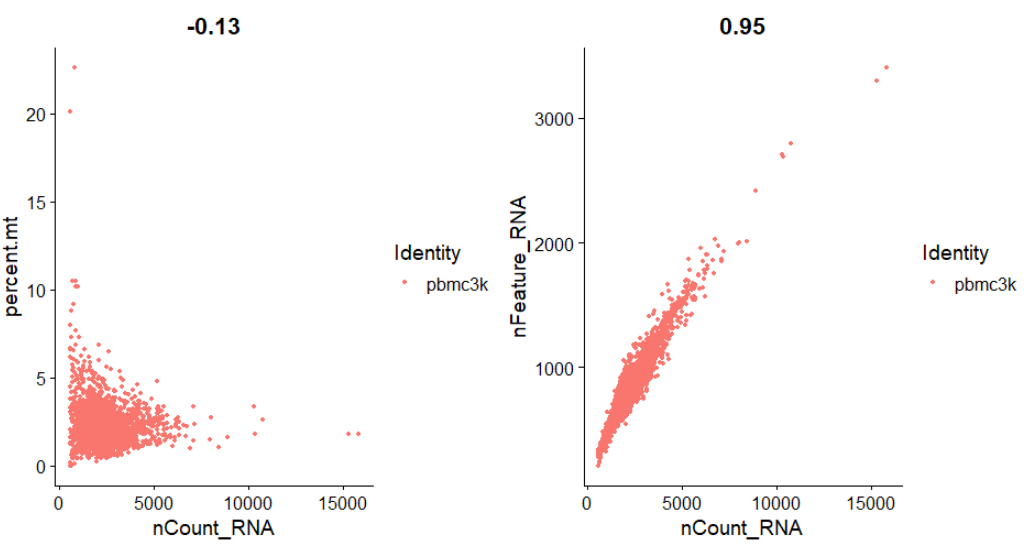
3.2 三个指标之间的相关性,正常是正比例,不太重要
plot1 <- FeatureScatter(pbmc,
feature1 = "nCount_RNA",
feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc,
feature1 = "nCount_RNA",
feature2 = "nFeature_RNA")
plot1 + plot2
# 过滤
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
pbmc
4. 标准化,找表达高变基因(HVG)
## =============4.标准化
pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize",
scale.factor = 10000) #先标准化数据 消除不同细胞测序深度的影响
# 标准化后的值保存在:pbmc[["RNA"]]@data
normalized.data <- pbmc[["RNA"]]@data
normalized.data[1:20,1:4]
dim(normalized.data)
## =============5.鉴定高变基因
# 高变基因:在一些细胞中表达高,另一些细胞中表达低的基因
# 变异指标: mean-variance relationship
# 默认返回两千个高变基因,用于下游如PCA降维分析
pbmc <- FindVariableFeatures(pbmc) # 筛选出变化比较大的基因,筛选标准是方差,默认2000个
top10 <- head(VariableFeatures(pbmc), 10);top10 #这里选了2000个,把前十个在图上标记出来。
plot1 <- VariableFeaturePlot(pbmc)
plot2 <- LabelPoints(plot = plot1,
points = top10,
repel = TRUE) # 有标记点的图
plot1 + plot2
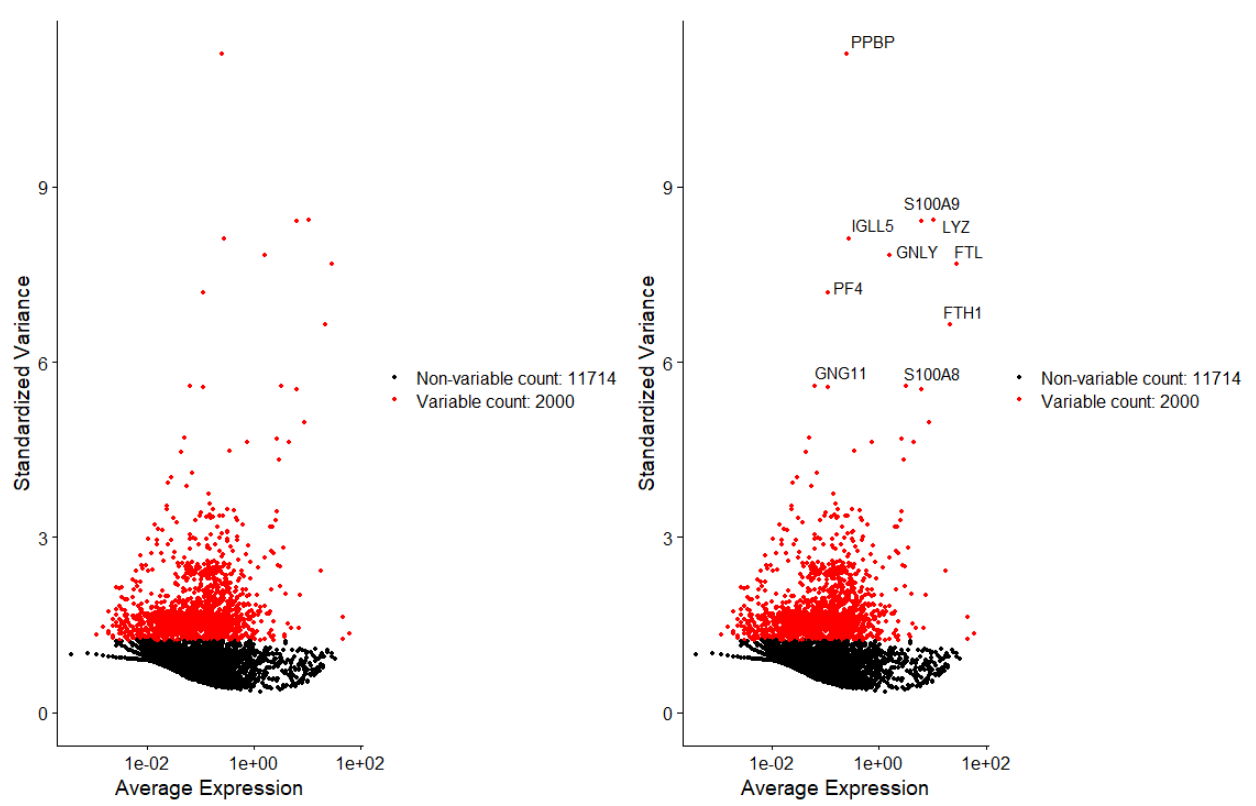
- 先标准化数据 NormalizeData(pbmc) 消除不同细胞测序深度的影响 降低数据离散程度 参考: https://www.jianshu.com/p/4a15cf570b87
- FindVariableFeatures(),筛选出变化比较大的基因,筛选标准是标准差,默认2000个
- VariableFeaturePlot()可视化,标准差和表达量, 2000个细胞
5 标准化和降维
#### 归一化 all.genes <- rownames(pbmc) # 基因名 pbmc <- ScaleData(pbmc, features = all.genes) # 添加所有基因归一化处理 表达量进行z-score转换 pbmc[["RNA"]]@scale.data[30:34,1:3]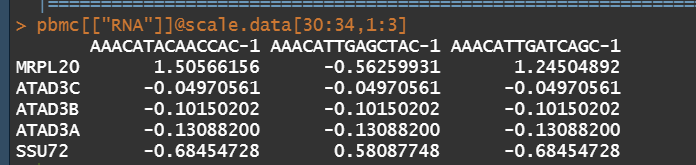
- ScaleData()添加归一化处理属性 将基因表达量的数值进行了z-score的转换 正态化处理
- PCA分析需要输入 正态化的数据
- 在 pbmc@assays[[“RNA”]]@scale.data 查看scale后的数据
- scale.data 有负值
5.1 线性降维PCA
```r pbmc <- RunPCA(pbmc, features = VariableFeatures(pbmc)) #只使用变异较大的基因来降维
查看前2个主成分由哪些feature组成
print(pbmc[[“pca”]], dims = 1:2, nfeatures = 30) VizDimLoadings(pbmc, dims = 1:2, reduction = “pca”)
每个主成分对应基因的热图
DimHeatmap(pbmc, dims = 1:2, cells = 500) # 选500个细胞来画热图
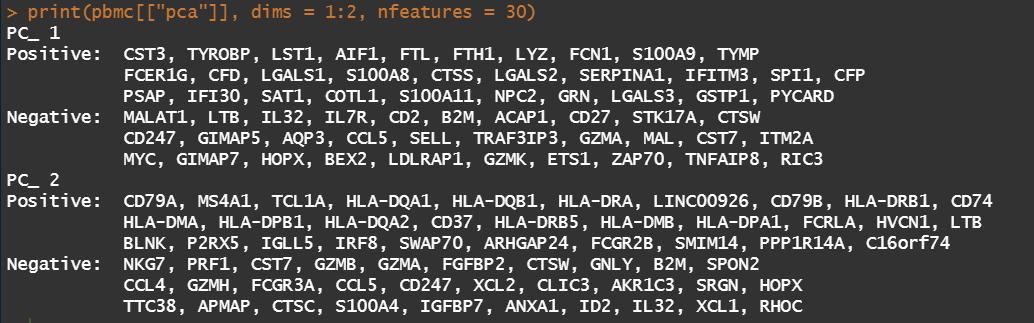<br />查看dim=2的主成分PCA nfeature=30*2<br />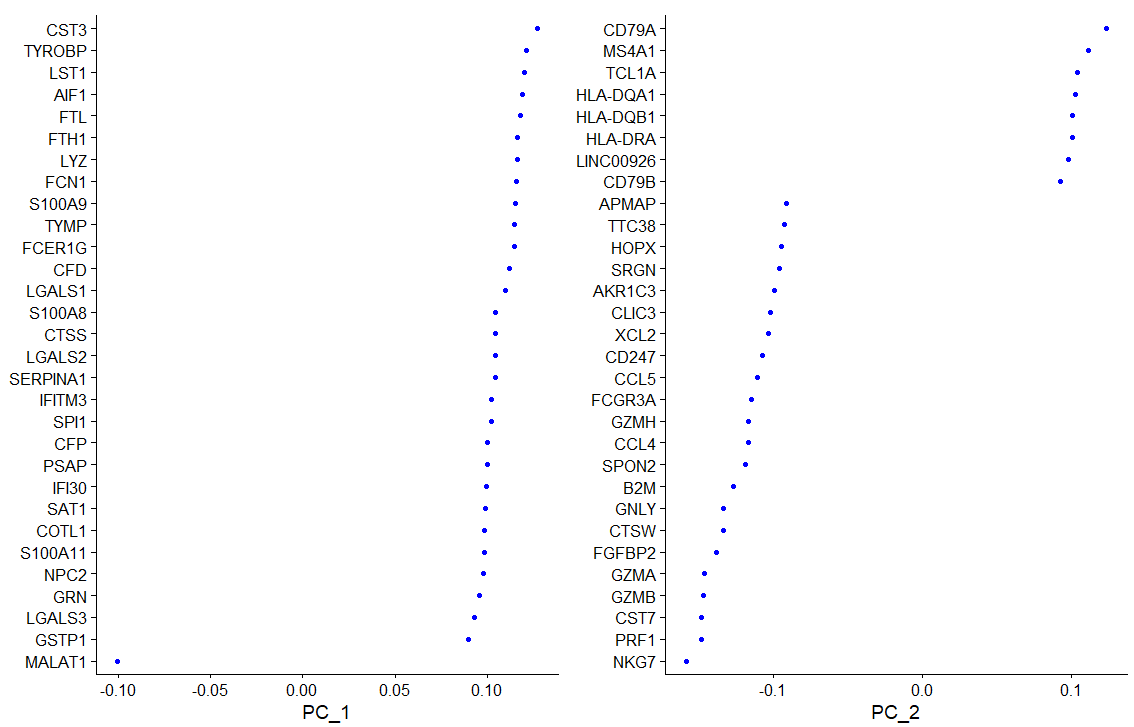<br />可视化查看前dim=2时的主成分,默认nfeature=30<br />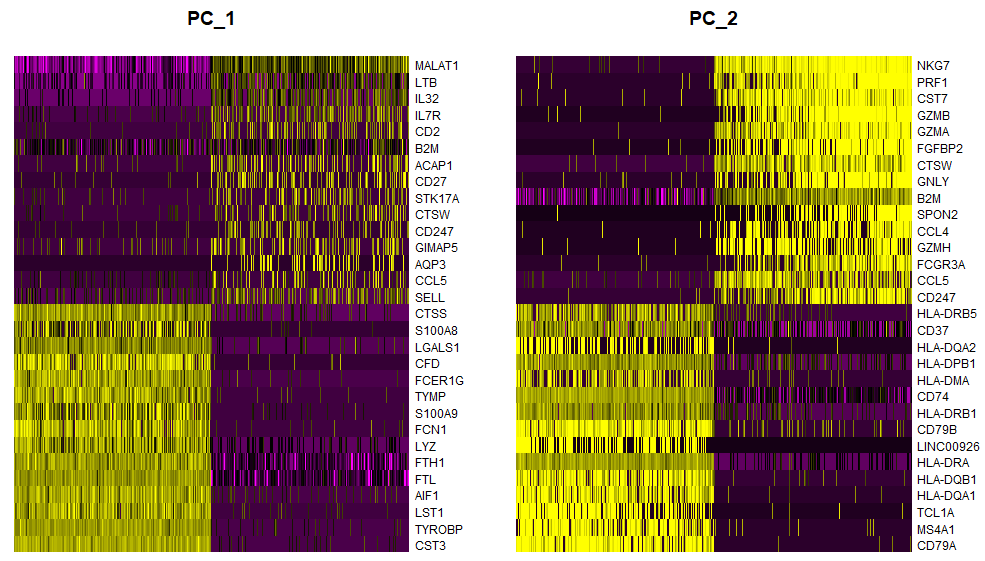<br />可视化查看前dim=2时的主成分,默认nfeature=30
应该选dim=多少呢?才是最佳降维呢?ElbowPlot(pbmc) JackStraw()
```r
# 应该选多少个主成分进行后续分析
ElbowPlot(pbmc)
pbmc <- JackStraw(pbmc, num.replicate = 100)
pbmc <- ScoreJackStraw(pbmc, dims = 1:20)
# 方式1:根据p值判断
JackStrawPlot(pbmc, dims = 1:15)
# 方式2:肘部法,根据拐点判断,比较主管,尽量多选几个PC
ElbowPlot(pbmc, ndims = 30)
# 结合JackStrawPlot和ElbowPlot,挑选10个PC
# PC1和2
# DimPlot(pbmc, reduction = "pca")+ NoLegend()
JackStrawPlot: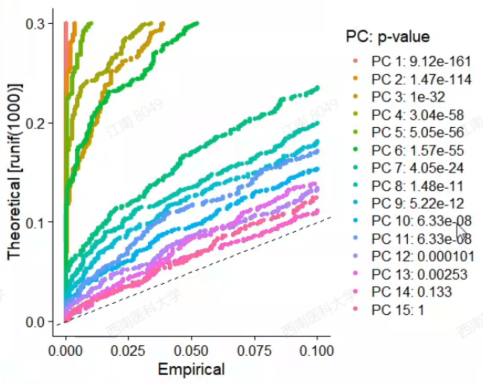
ElbowPlot: ElbowPlot(pbmc)看拐点, 默认20个pca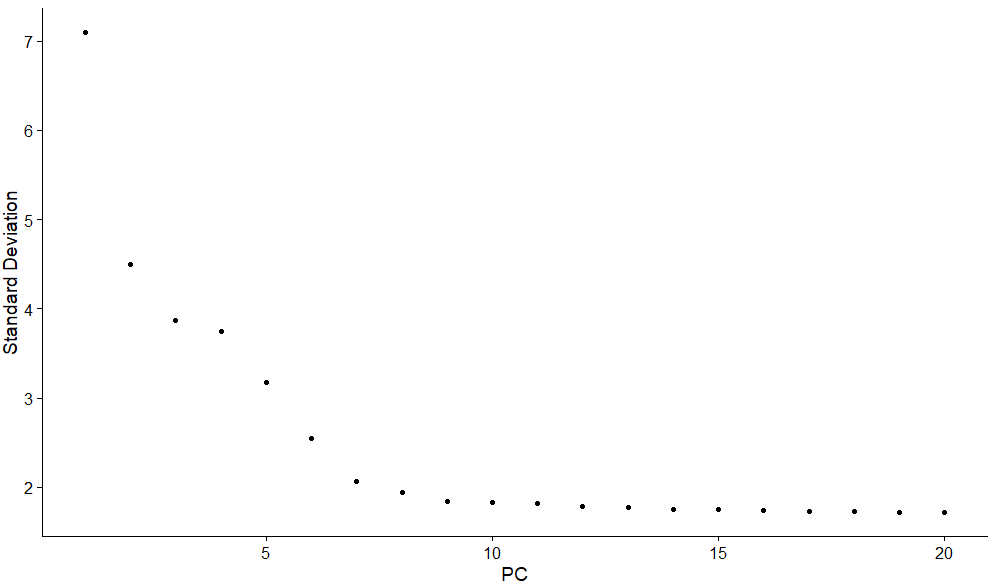
JackStrawPlot(pbmc, dims = 1:20)
5.2 聚类
## =============对细胞聚类
# 结合JackStrawPlot和ElbowPlot,挑选10个PC,所以这里dims定义为1:10
pbmc <- FindNeighbors(pbmc, dims = 1:10) # 找相似细胞
pbmc <- FindClusters(pbmc, resolution = 0.5) #聚类 分辨率 越大越多组---------------------
# 结果聚成几类,用Idents查看
length(levels(Idents(pbmc))) # 读取active.ident
# 9
# resolution对聚类的影响
res.used <- seq(0.1,1,by=0.2)
res.used
for(i in res.used){
pbmc <- FindClusters(pbmc, verbose = T, resolution = res.used)
}
# 可视化
library(clustree)
clus.tree.out <- clustree(pbmc) +
theme(legend.position = "bottom") +
scale_color_brewer(palette = "Set1") +
scale_edge_color_continuous(low = "grey80", high = "red")
clus.tree.out
5.3 UMAP 和t-sne 聚类
PCA是线性降维,这两个是非线性降维。
pbmc <- RunUMAP(pbmc, dims = 1:10) #输入为 pca降维数据
# 可视化
DimPlot(pbmc, reduction = "umap", label = T, label.size = 5)
DimPlot(pbmc, reduction = "tsne", label = T, label.size = 5)
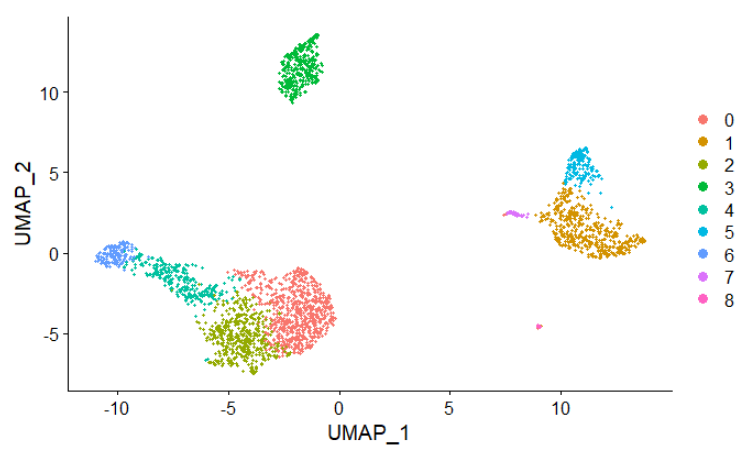
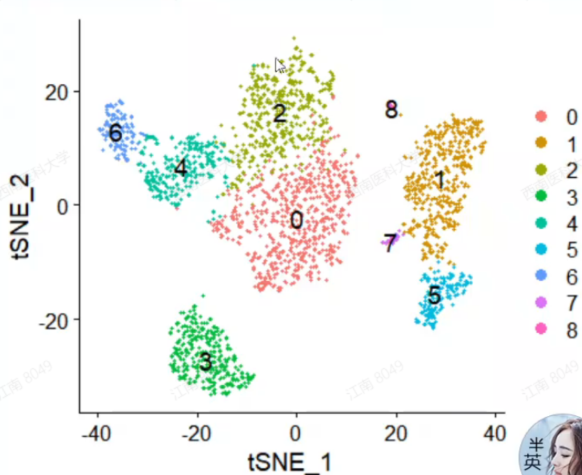
6.找marker基因
marker基因:和差异基因里面的上调基因有点类似,某个基因在某一簇细胞里表达量都很高,在其他簇表达量很低,那么这个基因就是这簇细胞的象征。
- 找全部cluster的maker基因
```r
=============差异表达分析
在cluster2 vs else中差异表达 (默认秩和检验)
cluster1.markers <- FindMarkers(pbmc, ident.1 = 2, min.pct = 0.25) head(cluster1.markers, n = 5)指定两个类cluster 5 from clusters 0 and 3
cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25) head(cluster5.markers, n = 5)
所有类的差异表达基因
only.pos:只保留上调差异表达的基因
pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, # 只返回positive基因 min.pct = 0.25)
只计算至少在(两簇细胞总数的)25%的细胞中有表达的基因
pbmc.markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_log2FC) # 每簇显示2个marker基因
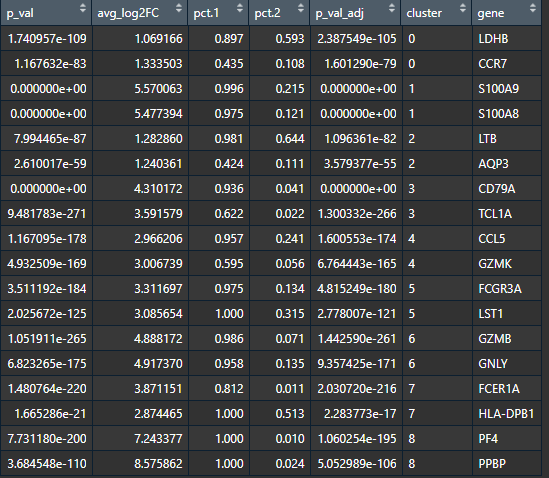
<a name="OF6qf"></a>
#### 6.1 比较某个基因在几个cluster之间的表达量
```r
#小提琴图
#可以拿count数据画
VlnPlot(pbmc, features = c("NKG7", "PF4"), slot = "counts", log = TRUE)
#在umap图上标记
FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A",
"FCGR3A", "LYZ", "PPBP", "CD8A"))
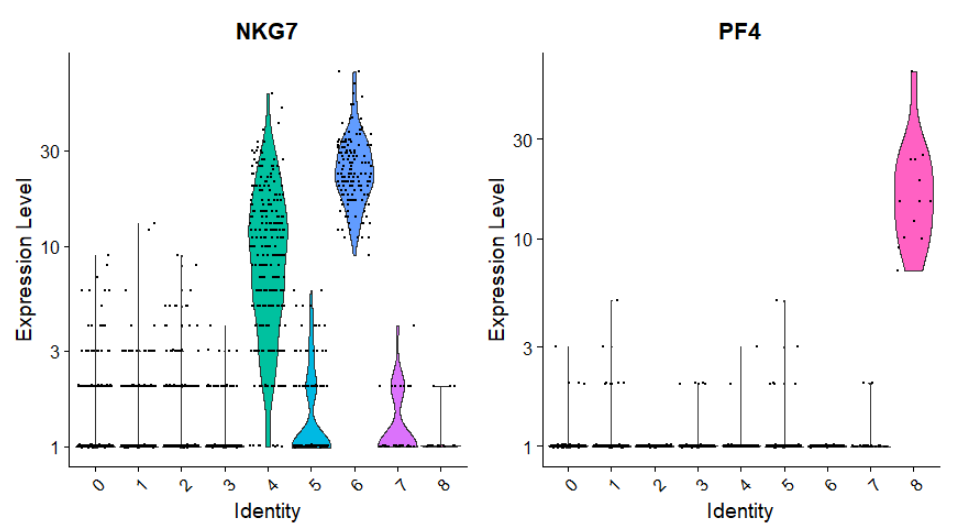
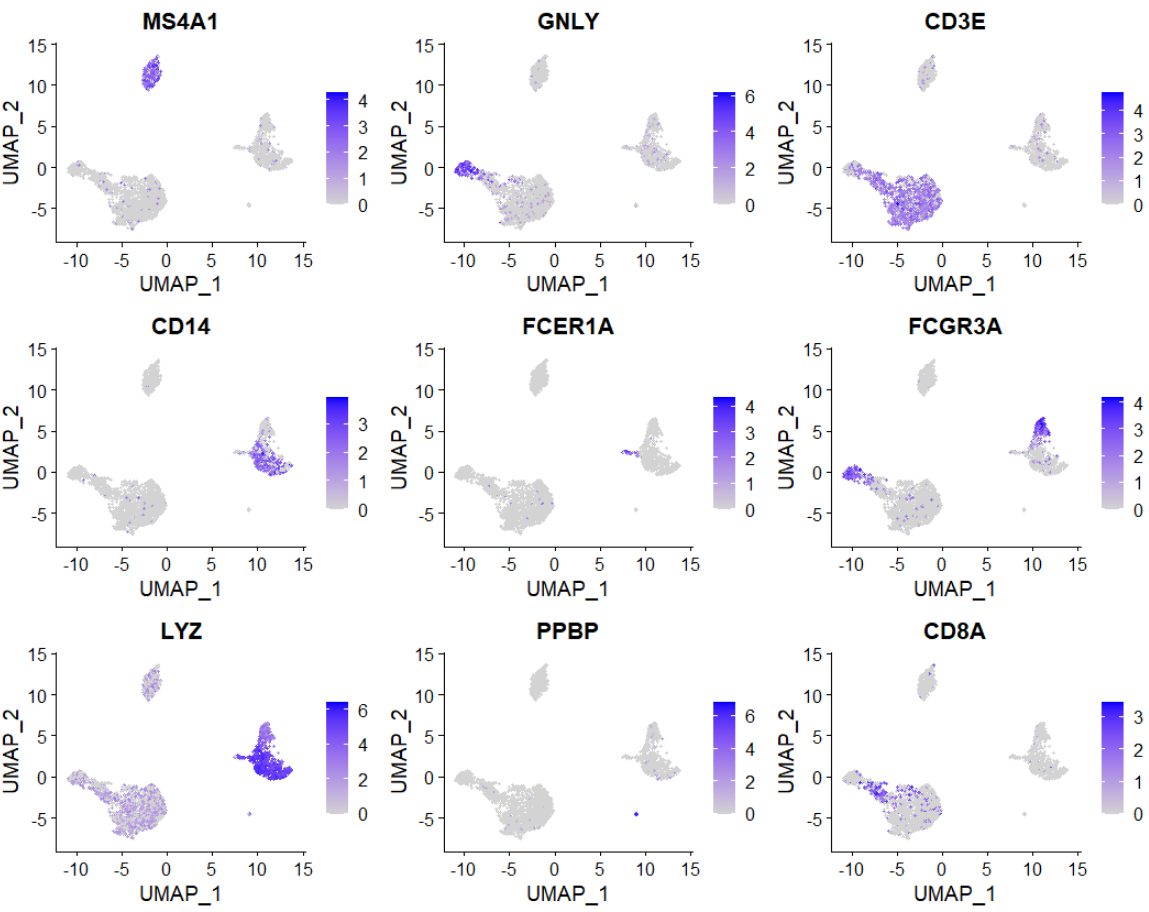
6.2 marker基因的热图 每簇top10 marker gene
top10 <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_log2FC)
DoHeatmap(pbmc, features = top10$gene)
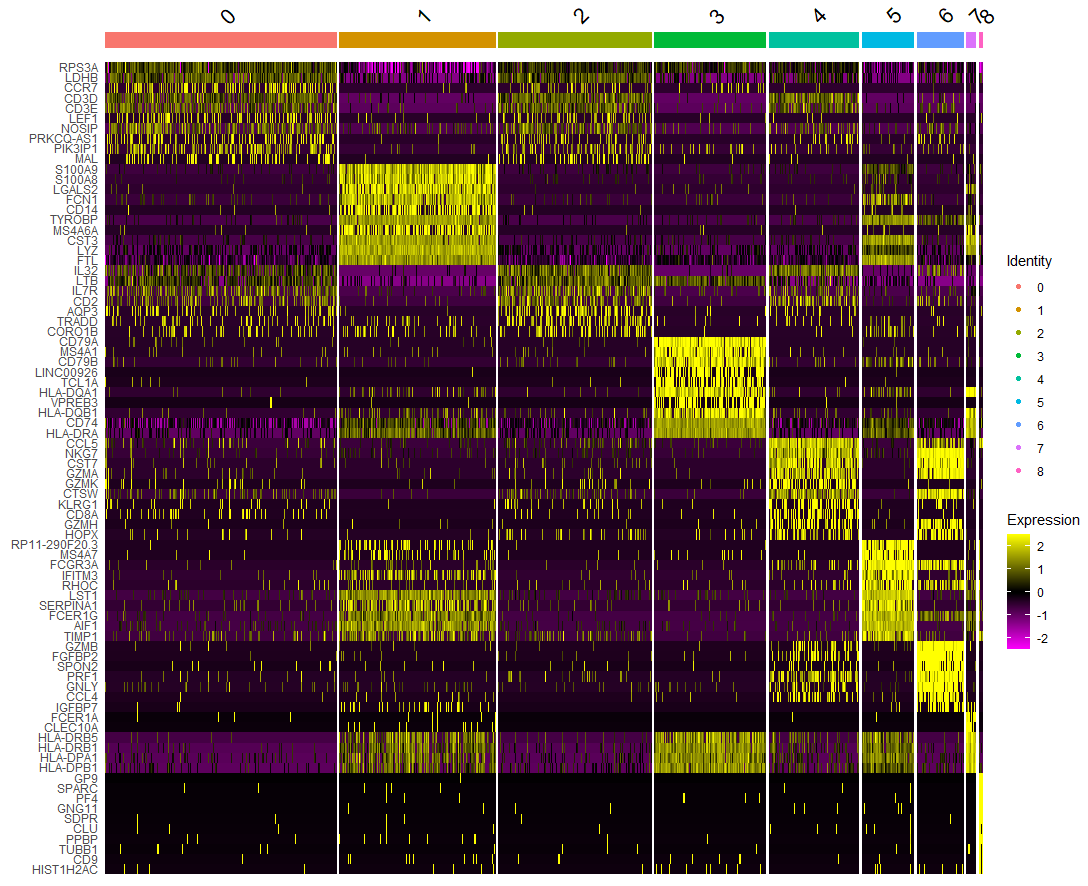
7. 根据marker基因确定细胞
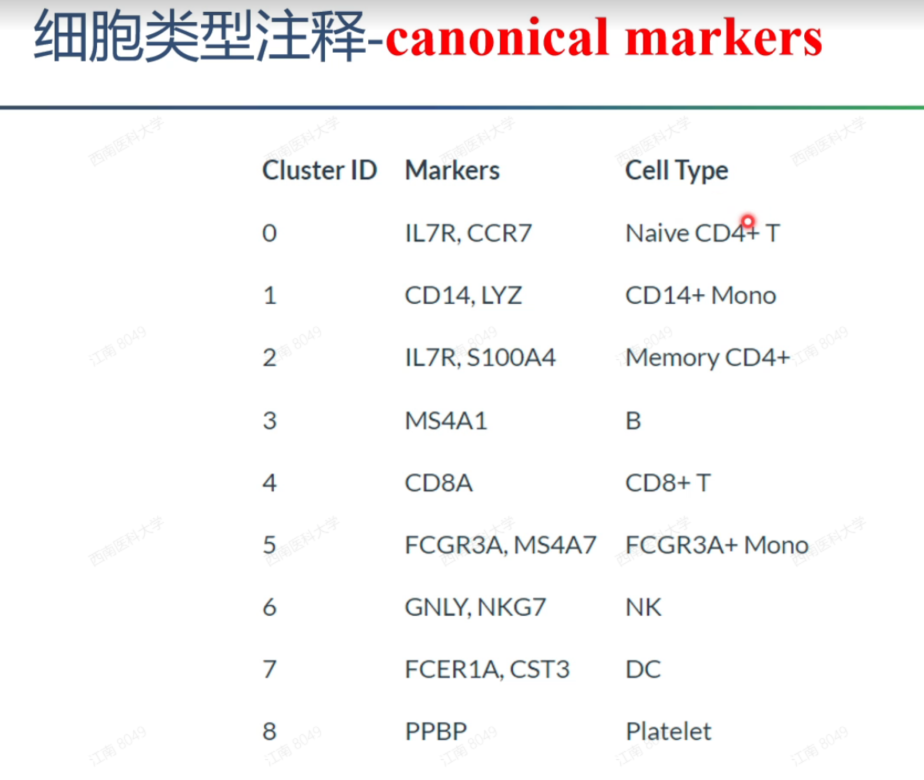
根据背景知识,也有R包 celldex 和 SingleR 根据marker基因 注释细胞
new.cluster.ids <- c("Naive CD4 T",
"CD14+ Mono",
"Memory CD4 T",
"B",
"CD8 T",
"FCGR3A+ Mono",
"NK",
"DC",
"Platelet")
names(new.cluster.ids) <- levels(pbmc)
pbmc <- RenameIdents(pbmc, new.cluster.ids)
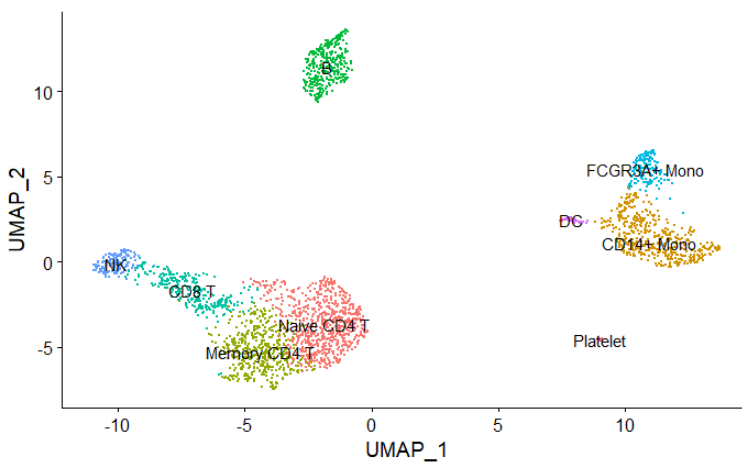
DimPlot(pbmc,
reduction = "umap",
label = TRUE,
pt.size = 0.5) + NoLegend()

若有收获,就点个赞吧
0 人点赞
