序列相似性
相似的序列往往起源于一个共同的祖先序列。它们很可能有相似的空间结构和生物学功能,因此对于一个已知序列但未知结构和功能的蛋白质,如果与它序列相似的某些蛋白质的结构和功能已知,则可以推测这个未知结构和功能的蛋白质的结构和功能。
一致度(identity):如果两个序列(蛋白质或核酸)长度相同,那么它们的一致度定义为他们对应位置上相同的残基(一个字母,氨基酸或碱基)的数目占总长度的百分数。
相似度(similarity):如果两个序列(蛋白质或核酸)长度相同,那么它们的相似度定义为他们对应位置.上相似的残基与相同的残基的数目和占总长度的百分数。
替换记分矩阵(Substitution Matrix):反映残基之间相互替换率的矩阵,它描述了残基两两相似的量化关系。分为DNA替换记分矩阵和蛋白质替换记分矩阵。
DNA序列替换记分矩阵:
- 等价矩阵(unitary matrix):最简单的替换记分矩阵,其中,相同核寄酸之间的匹配得分为 1,不同核苷酸间的替换得分为 0。由于不含有碱基的理化信息和不区别对待不同的替换,在实际的序列比较中较少使用。
- 转换-颠换矩阵( transition transversion matrix) :核酸的碱基按照环结构特征被划分为两类,一类是嘌呤(腺嘌呤A、鸟嘌呤G),它们有两个环;另一类是嘧啶(胞嘧啶C、胸腺嘧啶T),它们只有一个环。如果DNA碱基的替换保持环数不变,则成为转换,如A→G、C→T;如果环数发生变化,则成为颠换,如A→C、T→G等。在进化过程中,转换发生的频率远比颠换高。为了反映这一情况,通常该矩阵中转换的得分为-1,而颠换的得分为-5。
- BLAST矩阵:经过大量实际比对发现,如果令被比对的两个核苷酸相同时得分为+5,反之为-4,则比对效果较好。这个矩阵广泛地被DNA序列比较所采用。
蛋白质替换记分矩阵:
- 等价矩阵(unitarymatrix) :与DNA等 价矩阵道理相同,相同氨基酸之间的匹配得分为1,不同氨基酸间的替换得分为0。在实际的序列比对中较少使用。
- PAM矩阵(Dayhoff突 变数据矩阵) : PAM矩阵基于进化原理。如果两种氨基酸替换频繁,说明自然界易接受这种替换,那么这对氨基酸替换得分就应该高。PAM矩阵是目前蛋白质序列比较中最广泛使用的记分方法之一,基础的PAM-1矩阵反应的是进化产生的每一百个氨基酸平均发生一个突变的量值(统计方法得到)。PAM-1自乘n次,可以得到PAM-n,即发生了更多次突变。
- BLOSUM矩阵(blocks substitution matrix) : BLOSUM矩阵都是通过对大量符合特定要求的序列计算而来的。PAM-1矩阵是基于相似度大于85%的序列计算产生的,那些进化距离较远的矩阵,如PAM-250,是通过PAM-1自乘得到的。即 BLOSUM矩阵的相似性是根据真实数据产生的,而PAM矩阵是通过矩阵自乘外推而来的。BLOSUM矩阵的编号,比如BLOSUM-80中的80,代表该矩阵是由一致度≥80%的序列计算而来的,同理,BLOSUM-62是 指该矩阵由一致度≥62% 的序列计算而来的。


对于关系较远的序列之间的比较,由于PAM-250是推算而来,所以其准确度受到一定限制,BLOSUM-45更具优势。对于关系较近的序列之间的比较,用PAM或BLOSUM矩阵做出的比对结果,差别不大。最常用的: BLOSUM- 62
- 遗传密码矩阵(genetic code matrix, GCM) :遗传密码矩阵通过计算一个氨基酸转换成另一个氨基酸所需的密码子变化的数目而得到,矩阵的值对应为据此付出的代价。如果变化一个碱基就可以使一个氨基酸的密码子转换为另一个氨基酸的密码子,则这两个氨基酸的替换代价为1;如果需要2个碱基的改变,则替换代价为2;再比如从Met到Tyr三个密码子都要变,则代价为3。遗传密码矩阵常用于进化距离的计算,其优点是计算结果可以直接用于绘制进化树,但是它在蛋白质序列比对(尤其是相似程度很低的蛋白质序列比对)中,很少被使用。
- 疏水矩阵:根据氨基酸残基替换前后疏水性的变化而得到得分矩阵。若一次氨基酸替换疏水特性不发生太大的变化,则这种替换得分高,否则替换得分低
短串联重复序列( short tandem repeat, STR)也叫做微卫星DNA,是一类广泛存在于真核生物基因组中的DNA串联重复序列。它由2-6bp的核心序列组成,重复次数通常在15-30次。STR具有高度多态性,即存在重复次数的个体间差异,而且这种差异在基因遗传过程中一般遵循孟德尔共显性遗传规律,所以它被广泛用于法医学个体识别、亲子鉴定等领域。
Needleman -Wunsch Algorithm
Matching Sequences under Deletion/Insertion Constraints
:::warning 书本上的案例是原论文的翻译,。现在网上能搜索到的动态规划版本不是原论文的状态转移方程。
如果不懂动态规划这一节也不太可能考,建议先学习下暴力枚举,尤其是回溯函数(八皇后)
算法具体细节见下面的文档,来源于网络,下面只给出实现思考细节。
:::
在介绍下面代码实现思路之前,先推一个供参考学习工具,该作者实现在线网站供显示双序列全局比对的结果展示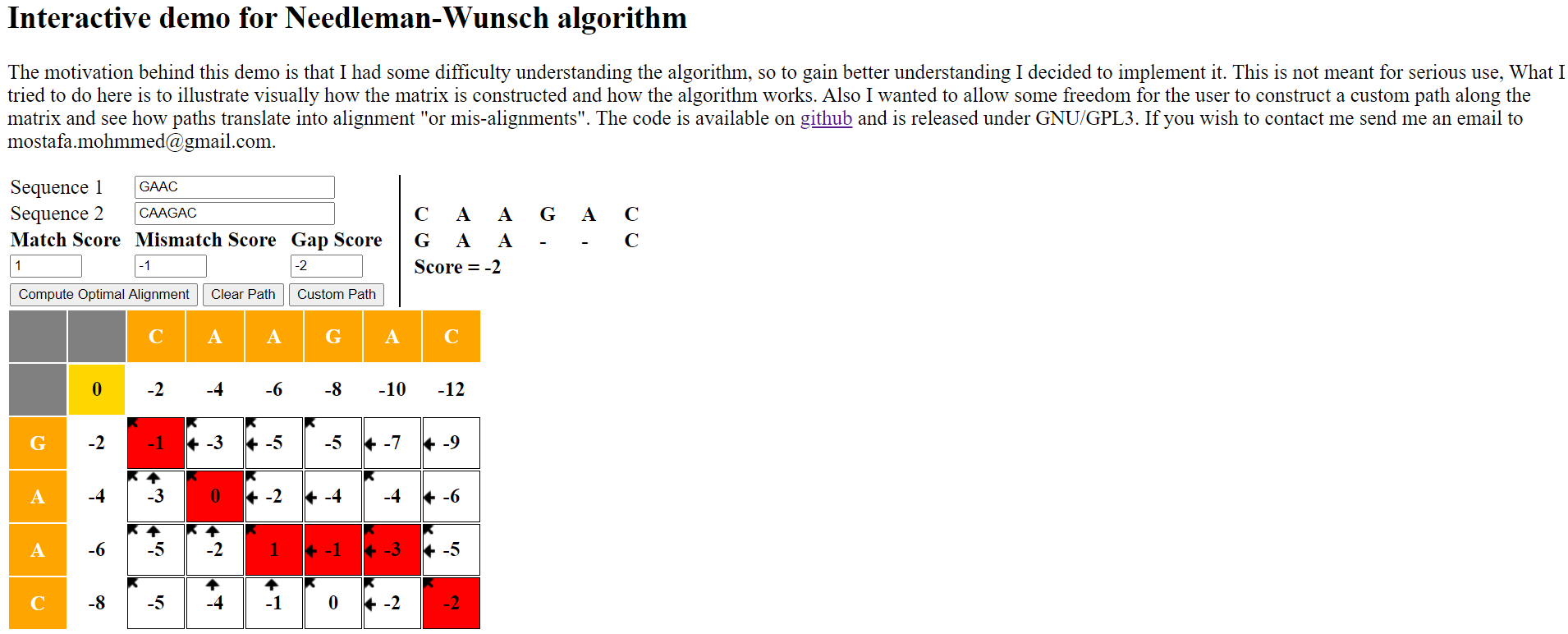
该工具使用很方便,使用 JavaScript 编写的网页,同时集成较好的交互,非常适于可视化学习,其在线版本由于某些文件加载可能需要科学上网,因此此处已将其在 Github 发布下载到本地。直接用浏览器打开 index.html 即可。Needleman-Wunsch-master.zip
1. BF 算法(自顶向下:穷举所有可能)
对于每对氨基酸比较,一共有两种可能:匹配或者不匹配。但是不匹配又有两种选择:①不加入空位 ②加入空位(那条序列加入呢?还是两条都加?)。
因此一共如下几种可能:
- 完全匹配:例如
'A' = 'A' 不匹配:
- 不插入空位,算这位蒙混过关,但是要扣分!例如
'ABC' = 'ADC',次数若在A-DC插入空位会导致后面更加失配。 插入空位(gap),扣分力度最大!例如
'ABC' = 'AC',显然后者不够全局匹配必须插入空位'A-C'才能满足。注意两条序列中任一条都可插入空位。 :::tips Q:为什么没有不匹配的时候两条序列都插入空位呢?如'AC' & 'BC',为何不在第一个都插入空位呢?
A: 两者同时插入空位是没有任何意义的!'-AC' & '-BC'。这不仅没有任何生物学意义,而且带来的罚分是双倍的。 ::: 因此根据需求可以写出最暴力的回溯枚举代码,每个比对位置含有三种状态选择。 ```python class NW: def init(self, match=1, mismatch=-1, gap=-2) -> None: “””For each alignment, three options to operate””” self.match = match self.mismatch = mismatch self.gap = gapdef NeedlemanWunsch(self, seq1: str, seq2: str) -> int: n, m = len(seq1), len(seq2) self.score = float(‘-inf’) def solve(idx1: int, idx2: int, score: int) -> None:
if idx1 >= n and idx2 >= m:self.score = max(self.score, score)return# 1.match/matchif idx1 < n and idx2 < m:solve(idx1 + 1, idx2 + 1,score + (self.match if seq1[idx1] == seq2[idx2] else self.mismatch))# 2.insert gap in seq1if idx2 < m:solve(idx1, idx2 + 1, score + self.gap)# 3.insert gap in seq2if idx1 < n:solve(idx1 + 1, idx2, score + self.gap)
solve(0, 0, 0) return self.score
- 不插入空位,算这位蒙混过关,但是要扣分!例如
s = NW() seq1 = “TACTCG” seq2 = “AATCAC” print(s.NeedlemanWunsch(seq1, seq2)) # -3
下面会爆炸!
seq1 = “ABCNJRQCLCRPM”
seq2 = “AJCJNRCKCRBP”
print(s.NeedlemanWunsch(seq1, seq2))
上面这种是最容易想到的暴力枚举方式,每个状态都传递下当前的值,但是有一个问题就是如何知道最后的解决办法呢??假设有多个最优解呢?也就是路径记录问题。使用全局路径数组进行记录即可。参考代码如下,每次进行三个状态选择时,都要进行**撤销**选择状态。```pythonfrom typing import Listclass NW:def __init__(self, match=1, mismatch=-1, gap=-2) -> None:"""For each alignment, three options to operate"""self.match = matchself.mismatch = mismatchself.gap = gapdef NeedlemanWunsch(self, seq1: str, seq2: str) -> List[str]:n, m = len(seq1), len(seq2)self.score = float('-inf')path1, path2 = [], []res = []def solve(idx1: int, idx2: int, score: int) -> None:if idx1 >= n and idx2 >= m:if score >= self.score:if score > self.score:res.clear() # 清空之前的答案res.append([score, ''.join(path1), ''.join(path2)])self.score = scorereturn# 1.match/matchif idx1 < n and idx2 < m:path1.append(seq1[idx1])path2.append(seq2[idx2])solve(idx1 + 1, idx2 + 1,score + (self.match if seq1[idx1] == seq2[idx2] else self.mismatch))path1.pop()path2.pop()# 2.insert gap in seq1if idx2 < m:path1.append('-')path2.append(seq2[idx2])solve(idx1, idx2 + 1, score + self.gap)path1.pop()path2.pop()# 3.insert gap in seq2if idx1 < n:path2.append('-')path1.append(seq1[idx1])solve(idx1 + 1, idx2, score + self.gap)path1.pop()path2.pop()solve(0, 0, 0)return ress = NW()seq1 = "TACTCG"seq2 = "AATCAC"for score, s1, s2 in s.NeedlemanWunsch(seq1, seq2):print(score)print(s1)print(s2)print()
输出结果:多个最优结果进行打包。
-3TA-CTCGAATCAC--3TACTCG-AA-TCAC-3TACTC-GAA-TCAC-3TACTCG--AATCAC-3TACTC-G-AATCAC
复杂度分析:以  分别代表两序列的长度。
分别代表两序列的长度。
- 时间复杂度:
 ,对于一条序列上一个氨基酸,它有两中可能,插入空位或不插入空位(包含不匹配+匹配)。
,对于一条序列上一个氨基酸,它有两中可能,插入空位或不插入空位(包含不匹配+匹配)。 - 空间复杂度:,递归树的高度!
2. 记忆化递归
上述代码比较直接容易理解,但是很容易发现其实它进行很多次重复的递归。例如:
在某次比对中,假设此时idx1 = 0, idx2 = 0,可以选择
- 不插入空位,两序列开始下一个匹配;
 插入空位;
插入空位; 插入空位;
插入空位;
若此时选择 1 操作,即 solve(idx1 + 1, idx2 + 1, score + self.gap) , 那么此后需要的比对分值来自  的结果加上之前比对结果总和。
的结果加上之前比对结果总和。
若此时选择 2 操作,即 solve(idx1, idx2 + 1, score + self.gap) , 那么此后需要的比对分值来自  的结果加上之前比对结果总和。
的结果加上之前比对结果总和。
- 此时来到
idx1 = 0, idx2 = 1,同上三种选择,若选择3操作,即solve(idx1 + 1, idx2, score + self.gap), 那么此后需要的比对分值来自的结果加上之前比对结果总和。
若此时选择 3 操作,即 solve(idx1 + 1, idx2, score + self.gap) , 那么此后需要的比对分值来自  的结果加上之前比对结果总和。
的结果加上之前比对结果总和。
- 此时来到
idx1 = 1, idx2 = 0,同上三种选择,若选择2操作,即solve(idx1, idx2 + 1, score + self.gap), 那么此后需要的比对分值来自的结果加上之前比对结果总和。
综上举例说明,在某次选择操作 1 后,其之后的分值其实已经求出。重复求解地方如下:
- 当先选
2操作,到下一对比较时又选择3操作又要求解一次之后的分值。 - 当先选
3操作,到下一对比较时又选择2操作又要求解一次之后的分值。
因此我们尽可能要必要这种重复求解过程,这也是为什么递归做了很多没用功!
问题就是如何让计算机知道  已经求过了,不用再次递归求解了?很明显我们用数组把它们的值给记录下来,当递归函数再次调用前,我们先查数组内是否有记录过,有就直接返回,否则求解并把它的值再记录到数组中。
已经求过了,不用再次递归求解了?很明显我们用数组把它们的值给记录下来,当递归函数再次调用前,我们先查数组内是否有记录过,有就直接返回,否则求解并把它的值再记录到数组中。
参考代码:
python 中有一个缓存装饰器函数,非常方便。但是需要修改函数设计,因为上面的代码中函数没有返回值,且即使 seq1,seq2 出现过,但是 score 参数也不一定相等!
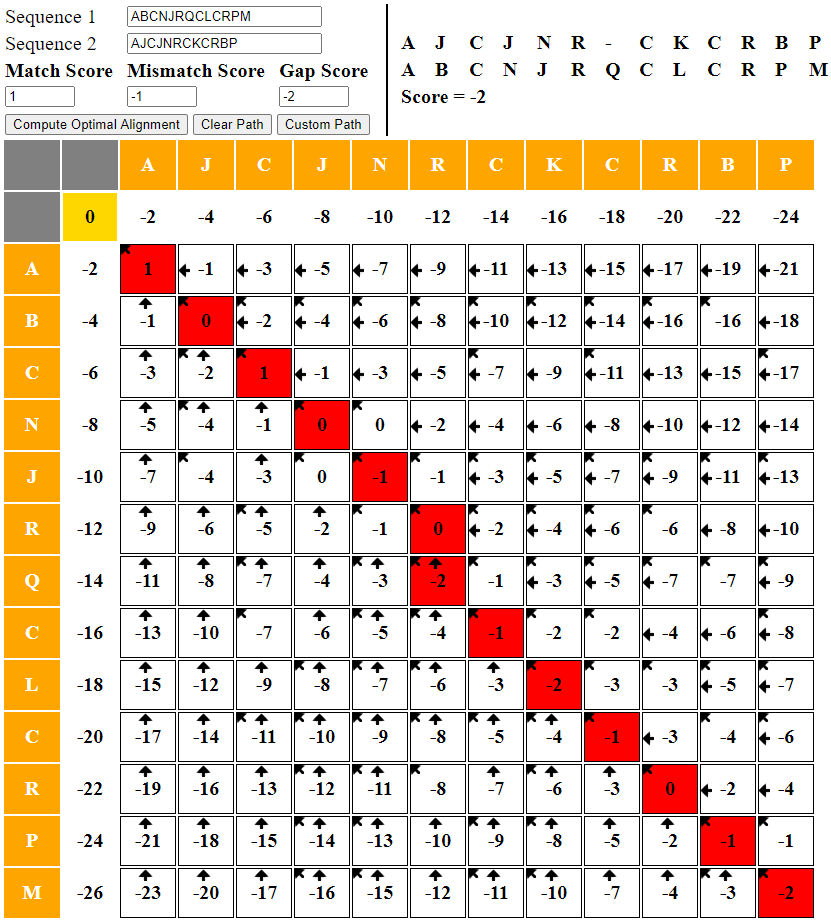
class NW:def __init__(self, match=1, mismatch=-1, gap=-2) -> None:"""For each alignment, three options to operate"""self.match = matchself.mismatch = mismatchself.gap = gapdef NeedlemanWunsch(self, seq1: str, seq2: str) -> int:m, n = len(seq1), len(seq2)from functools import lru_cache@lru_cache(None)def solve(idx1: int, idx2: int) -> int:if idx1 >= m or idx2 >= n: # 某个不足return min(0, idx1 - m) * self.gap + min(0, idx2 - n) * self.gapscore = float('-inf')# 1.matchif seq1[idx1] == seq2[idx2]:score = max(score, solve(idx1 + 1, idx2 + 1) + self.match)# 2.mismatchscore = max(score, solve(idx1 + 1, idx2 + 1) + self.mismatch)# 3.insert gap in seq1score = max(score, self.gap + solve(idx1, idx2 + 1))# 3.insert gap in seq2score = max(score, self.gap + solve(idx1 + 1, idx2))return scorereturn solve(0, 0)s = NW()seq1 = "ABCNJRQCLCRPM"seq2 = "AJCJNRCKCRBP"print(s.NeedlemanWunsch(seq1, seq2)) # -2
我们用数组实现这个缓存机制,然后打印出所有的得分信息。
#include <stdio.h>#include <limits.h>#include <string.h>#include <math.h>#define MATCH 1#define MISMATCH -1#define GAP -2#define N 20#define MIN -2139062144char seq1[] = "ABCNJRQCLCRPM"; // BCNJRQCLCRPMchar seq2[] = "AJCJNRCKCRBP"; // JCJNRCKCRBPint memo[N][N]; // 初始化为 INT_MINint NeedlemanWunsch(int i, int j) {if (i < 0 || j < 0) { // 不够长return fmax(i, 0) * GAP + fmax(j, 0) * GAP;} else if (MIN != memo[i][j]) { // 访问过return memo[i][j];} else {// 1.matchif (seq1[i] == seq2[j]) {memo[i][j] = fmax(memo[i][j],NeedlemanWunsch(i - 1, j - 1) + MATCH);}// 2.mismatchmemo[i][j] = fmax(memo[i][j],NeedlemanWunsch(i - 1, j - 1) + MISMATCH);// 3.insert gap in seq1memo[i][j] = fmax(memo[i][j],GAP + NeedlemanWunsch(i, j - 1));// 4.insert gap in seq2memo[i][j] = fmax(memo[i][j],GAP + NeedlemanWunsch(i - 1, j));}return memo[i][j];}int main() {memset(memo, 0x80, sizeof(memo)); // 偷个懒,-2139062144int m = strlen(seq1), n = strlen(seq2);printf("Max score: %d\n", NeedlemanWunsch(m - 1, n - 1));printf(" ");for (int j = 0; j < n; j++) printf(" %c ", seq2[j]);printf("\n");for (int i = 0; i < m; i++) {printf("%c ", seq1[i]);for (int j = 0; j < n; j++) {printf("%3d ", memo[i][j]);}printf("\n");}return 0;}
编译运行:
b12@PC:~/bioInfo$ gcc -Wall ./src/needlemanWunsch.c -o ./bin/needlemanWunsch -lmb12@PC:~/bioInfo$ ./bin/needlemanWunschMax score: -2A J C J N R C K C R B PA 1 -1 -3 -5 -7 -9 -11 -13 -15 -17 -19 -21B -1 0 -2 -4 -6 -8 -10 -12 -14 -16 -16 -18C -3 -2 1 -1 -3 -5 -7 -9 -11 -13 -15 -17N -5 -4 -1 0 0 -2 -4 -6 -8 -10 -12 -14J -7 -4 -3 0 -1 -1 -3 -5 -7 -9 -11 -13R -9 -6 -5 -2 -1 0 -2 -4 -6 -6 -8 -10Q -11 -8 -7 -4 -3 -2 -1 -3 -5 -7 -7 -9C -13 -10 -7 -6 -5 -4 -1 -2 -2 -4 -6 -8L -15 -12 -9 -8 -7 -6 -3 -2 -3 -3 -5 -7C -17 -14 -11 -10 -9 -8 -5 -4 -1 -3 -4 -6R -19 -16 -13 -12 -11 -8 -7 -6 -3 0 -2 -4P -21 -18 -15 -14 -13 -10 -9 -8 -5 -2 -1 -1M -23 -20 -17 -16 -15 -12 -11 -10 -7 -4 -3 -2
3. 动态规划
我们将上述记忆化递归进行自底向上的递推过程,很明显递归函数的当前匹配来源于四个(“三个”)地方。
即状态转移方程:
- 状态初始化:
- 当两者都是空串,它们已经对齐,但是没有任何意义,因此设置状态

!seq1 || !seq2的其它情况下,很明显需要在较短串内插入空位才能全局对齐。
- 当两者都是空串,它们已经对齐,但是没有任何意义,因此设置状态

![2Y)DT)QVQ]G_EYK2`K0SEJ3.png](/uploads/projects/u1190503@gfsoma/2c238dcb08a195db550c792b8bd9b5e2.png)
- 开始填表:根据状态转移方程:对于
 主要由“三”个状态转移而来,分别是对角线、上方、右侧得到。
| dp[i-1][j-1] | dp[i-1][j] |
| —- | —- |
| dp[i][j-1] | dp[i][j] = max(dp[i-1][j-1] + isMatch, gap + dp[i-1][j], gap + dp[i][j-1]) |
主要由“三”个状态转移而来,分别是对角线、上方、右侧得到。
| dp[i-1][j-1] | dp[i-1][j] |
| —- | —- |
| dp[i][j-1] | dp[i][j] = max(dp[i-1][j-1] + isMatch, gap + dp[i-1][j], gap + dp[i][j-1]) |
#include <stdio.h>#include <limits.h>#include <string.h>#include <math.h>#define MATCH 1#define MISMATCH -1#define GAP -2#define N 20#define MIN -2139062144char seq1[] = "ABCNJRQCLCRPM"; // BCNJRQCLCRPMchar seq2[] = "AJCJNRCKCRBP"; // JCJNRCKCRBPint dp[N][N]; // 初始化为 INT_MINint NeedlemanWunsch(int m, int n) {// 1.状态初始化dp[0][0] = 0;for (int i = 1; i <= m; i++) {dp[i][0] = dp[i-1][0] + GAP;}for (int j = 1; j <= n; j++) {dp[0][j] = dp[0][j-1] + GAP;}// 2.状态转移for (int i = 1; i <= m; i++) {for (int j = 1; j <= n; j++) {dp[i][j] = fmax(dp[i-1][j-1] + (seq1[i-1] == seq2[j-1] ? MATCH : MISMATCH),fmax(dp[i-1][j] + GAP, dp[i][j-1] + GAP));}}return dp[m][n];}int main() {memset(dp, 0x80, sizeof(dp)); // 偷个懒,-2139062144int m = strlen(seq1), n = strlen(seq2);printf("Max score: %d\n", NeedlemanWunsch(m, n));printf(" ");for (int j = 0; j < n; j++) printf(" %c ", seq2[j]);printf("\n");for (int i = 0; i <= m; i++) {if (i) printf("%c ", seq1[i-1]);else printf(" ");for (int j = 0; j <= n; j++) {printf("%3d ", dp[i][j]);}printf("\n");}return 0;}
最终填完的表就是如下所示:
b12@PC:~/bioInfo$ gcc -Wall ./src/needlemanWunsch.c -o ./bin/needlemanWunsch -lmb12@PC:~/bioInfo$ ./bin/needlemanWunschMax score: -2Max score: -2A J C J N R C K C R B P0 -2 -4 -6 -8 -10 -12 -14 -16 -18 -20 -22 -24A -2 1 -1 -3 -5 -7 -9 -11 -13 -15 -17 -19 -21B -4 -1 0 -2 -4 -6 -8 -10 -12 -14 -16 -16 -18C -6 -3 -2 1 -1 -3 -5 -7 -9 -11 -13 -15 -17N -8 -5 -4 -1 0 0 -2 -4 -6 -8 -10 -12 -14J -10 -7 -4 -3 0 -1 -1 -3 -5 -7 -9 -11 -13R -12 -9 -6 -5 -2 -1 0 -2 -4 -6 -6 -8 -10Q -14 -11 -8 -7 -4 -3 -2 -1 -3 -5 -7 -7 -9C -16 -13 -10 -7 -6 -5 -4 -1 -2 -2 -4 -6 -8L -18 -15 -12 -9 -8 -7 -6 -3 -2 -3 -3 -5 -7C -20 -17 -14 -11 -10 -9 -8 -5 -4 -1 -3 -4 -6R -22 -19 -16 -13 -12 -11 -8 -7 -6 -3 0 -2 -4P -24 -21 -18 -15 -14 -13 -10 -9 -8 -5 -2 -1 -1M -26 -23 -20 -17 -16 -15 -12 -11 -10 -7 -4 -3 -2
复杂度分析:以 分别代表两序列的长度。
- 时间复杂度:
 ,对于一条序列上一个氨基酸,它有两中可能,插入空位或不插入空位(包含不匹配+匹配)。
,对于一条序列上一个氨基酸,它有两中可能,插入空位或不插入空位(包含不匹配+匹配)。 - 空间复杂度:,
dp表大小。4. 路径回溯
光得到分值还不够,如何把所有的匹配信息一一展示出来才是关键,如下图所示。 :::tips 在用浏览器打开index.html网页后,可以手动用鼠标取消某条路径,然后选择另一条最优路径(默认情况下它只展示一条最优路径)。 :::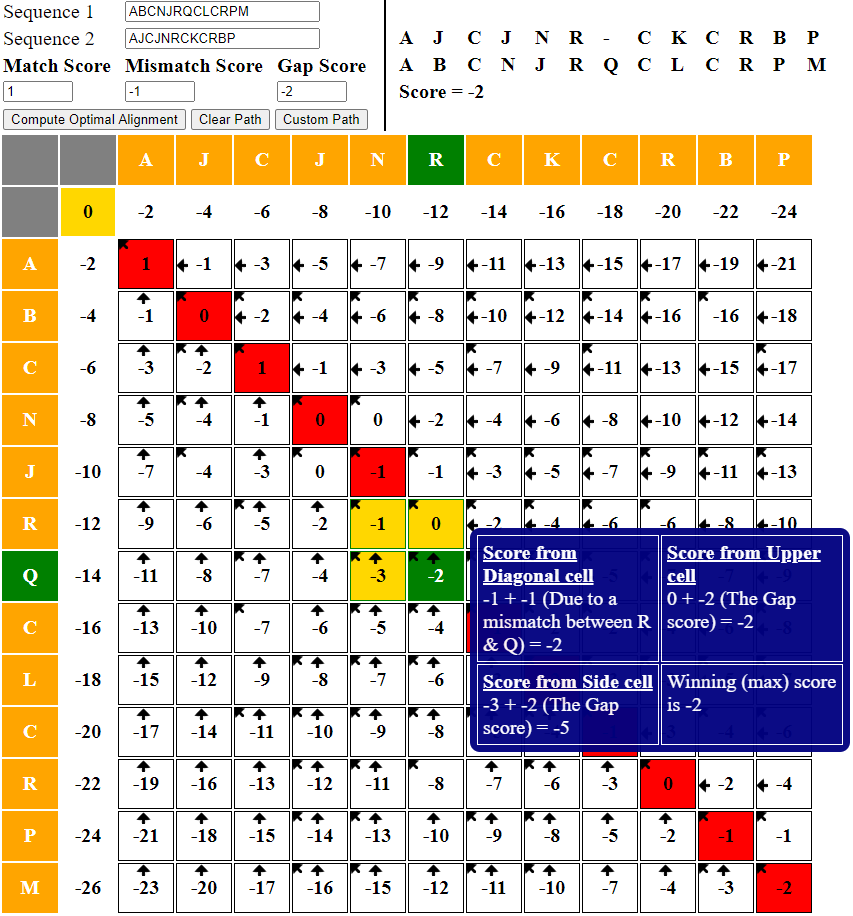
因此实现它就是从右下角回溯到左上角,根据状态转移方程(也可通过额外的标记数组实现)倒推回去。例如最右下角的-2此时 来自左对角线、上、左三个单元格的中的一个。
首先此时  ,
, ,两者不匹配,且此时
,两者不匹配,且此时  。分别根据转移方程进行如下三种倒推:
。分别根据转移方程进行如下三种倒推:
- 若来自上方,则是 插入空位,那么由
 得其上方理论值
得其上方理论值 ,但是其与实际记录值
,但是其与实际记录值 -1不符合。因此排除! - 若来自左方,则是 插入空位,那么由
 得其上方理论值
得其上方理论值 ,但是其与实际记录值
,但是其与实际记录值 -3不符合。因此排除! - 若来自左上方对角线,则有两种可能:
 ,不满足实际情况。注意:不能单纯判断
,不满足实际情况。注意:不能单纯判断  就是来自对角线!如下图左上角
就是来自对角线!如下图左上角 **-2 + 1**就是-1但是C != N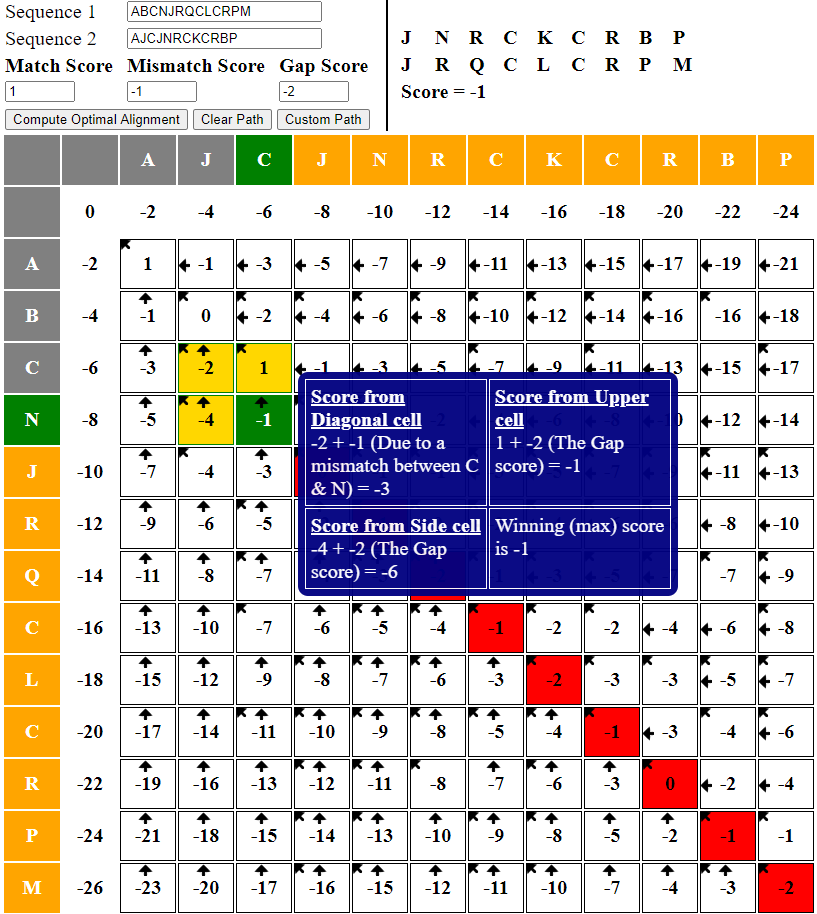
 ,则由
,则由  得
得  ,与实际记录值
,与实际记录值 -1符合。因此此时两字符不匹配,且最优子结构来自对角线!(下轮回溯到左上对角线继续开始判断)
综上所述,回溯的最终条件就是到到达左上角。见下方 PPT 展示
路径回溯.pptx
将其转化为代码(代码中只考虑一条最优结构)
class NW:def __init__(self, match=1, mismatch=-1, gap=-2) -> None:"""For each alignment, three options to operate"""self.match = matchself.mismatch = mismatchself.gap = gapdef NeedlemanWunsch(self, seq1: str, seq2: str) -> int:m, n = len(seq1), len(seq2)dp = [[0] * (n + 1) for _ in range(m + 1)]# 1. 状态初始化for i in range(1, m + 1):dp[i][0] = dp[i-1][0] + self.gapfor j in range(1, n + 1):dp[0][j] = dp[0][j-1] + self.gap# 2. 状态转移,注意下标for i in range(1, m + 1):for j in range(1, n + 1):dp[i][j] = max(dp[i-1][j-1] + (self.match if seq1[i-1] == seq2[j-1] else self.mismatch),dp[i-1][j] + self.gap,dp[i][j-1] + self.gap)# 3. 回溯输出匹配信息path1, path2, matchInfo = [], [], [] # 回溯路径def helper(i: int, j: int) -> None:if i + j == 0:print(' '.join(reversed(path2)))print(' '.join(reversed(matchInfo)))print(' '.join(reversed(path1)))print()returnif dp[i-1][j] == dp[i][j] - self.gap: # up:gap in seq2path1.append(seq1[i-1])matchInfo.append(' ')path2.append('-')helper(i - 1, j)elif dp[i][j-1] == dp[i][j] - self.gap: # left:gap in seq1path1.append('-')matchInfo.append(' ')path2.append(seq2[j-1])helper(i, j - 1)else: # diagonal:match/mismatchpath1.append(seq1[i-1])path2.append(seq2[j-1])if seq1[i-1] == seq2[j-1]:matchInfo.append('|')else:matchInfo.append('?') # mismatchhelper(i - 1, j - 1)helper(m, n)return dp[-1][-1]s = NW()seq1 = "ABCNJRQCLCRPM"seq2 = "AJCJNRCKCRBP"print('Max score:', s.NeedlemanWunsch(seq1, seq2)) # -2
运行结果:
A J C J N R - C K C R B P| ? | ? ? | | ? | | ? ?A B C N J R Q C L C R P MMax score: -2
:::tips 以上代码输出单条最优路径完全可以使用迭代实现,不需要使用递归(两者殊途同归)。本意只是想为后面通过递归函数回溯进行所有最优路径输出。(相比迭代实现所有最优路径输出,回溯函数代码更加简洁易懂) :::
5. 多条最优路径输出
我们从上面考点,在正中间 -2 地方,其实有两个方向。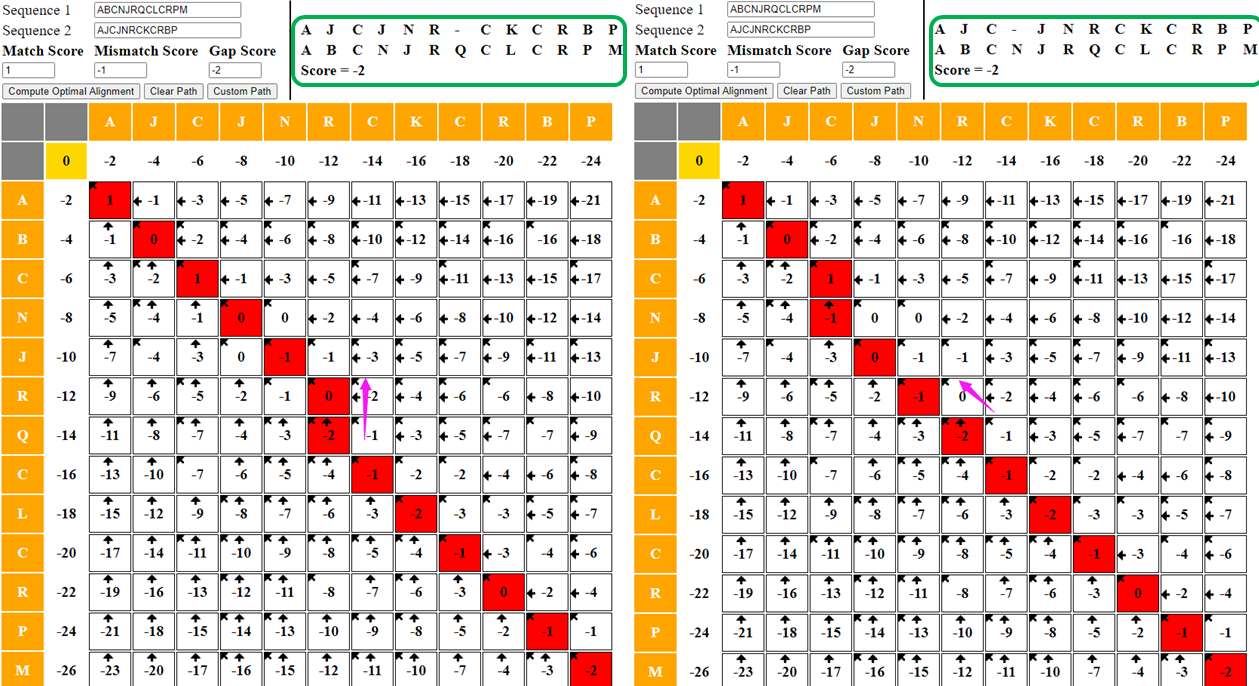 可供选择(向上: 插入一个
可供选择(向上: 插入一个  ;左上对角线:此时
;左上对角线:此时  是一个错配),由于我们代码逻辑是
是一个错配),由于我们代码逻辑是 if-elif-else 且考虑顺序是 up, left, diagonal ,因此在正中间 -2 地方位置选择向上回溯。
那么怎么才能实现把所有最优的结构打印出来呢?其实在正中间 -2 地方就是两个可选选择,要么向上,要么向左上角对角线去。因此就是两种状态的取舍啦(回溯最擅长枚举所有可能)。
- 将上面代码逻辑
if-elif-else改为if判断。 针对可能存在多个选择的路径时,我们需要对满足条件的挨个尝试,尝试一条路径到头后,里面回溯回来并撤销之前的选择,开始下一个选择。 ```python class NW: def init(self, match=1, mismatch=-1, gap=-2) -> None:
"""For each alignment, three options to operate"""self.match = matchself.mismatch = mismatchself.gap = gap
def NeedlemanWunsch(self, seq1: str, seq2: str) -> int:
m, n = len(seq1), len(seq2)dp = [[0] * (n + 1) for _ in range(m + 1)]# 1. 状态初始化for i in range(1, m + 1):dp[i][0] = dp[i-1][0] + self.gapfor j in range(1, n + 1):dp[0][j] = dp[0][j-1] + self.gap# 2. 状态转移,注意下标for i in range(1, m + 1):for j in range(1, n + 1):dp[i][j] = max(dp[i-1][j-1] + (self.match if seq1[i-1] == seq2[j-1] else self.mismatch),dp[i-1][j] + self.gap,dp[i][j-1] + self.gap)# 3. 回溯输出匹配信息path1, path2, matchInfo = [], [], [] # 回溯路径def helper(i: int, j: int) -> None:if i + j == 0:print(' '.join(reversed(path2)))print(' '.join(reversed(matchInfo)))print(' '.join(reversed(path1)))print()return# up:gap in seq2if dp[i-1][j] == dp[i][j] - self.gap:path1.append(seq1[i-1])matchInfo.append(' ')path2.append('-')helper(i - 1, j)path1.pop()matchInfo.pop()path2.pop()if dp[i][j-1] == dp[i][j] - self.gap:path1.append('-')matchInfo.append(' ')path2.append(seq2[j-1])helper(i, j - 1)path1.pop()matchInfo.pop()path2.pop()# diagonal:match(make sure seq1[i-1] == seq2[j-1])if seq1[i-1] == seq2[j-1] and dp[i-1][j-1] == dp[i][j] - self.match:path1.append(seq1[i-1])matchInfo.append('|')path2.append(seq2[j-1])helper(i - 1, j - 1)path1.pop()matchInfo.pop()path2.pop()# diagonal:mismatchif dp[i-1][j-1] == dp[i][j] - self.mismatch:path1.append(seq1[i-1])matchInfo.append('?')path2.append(seq2[j-1])helper(i - 1, j - 1)path1.pop()matchInfo.pop()path2.pop()helper(m, n)return dp[-1][-1]
s = NW() seq1 = “ABCNJRQCLCRPM” seq2 = “AJCJNRCKCRBP” print(‘Max score:’, s.NeedlemanWunsch(seq1, seq2)) # -2
**输出结果**:
A J C J N R - C K C R B P | ? | ? ? | | ? | | ? ? A B C N J R Q C L C R P M
A J C - J N R C K C R B P | ? | | ? ? | ? | | ? ? A B C N J R Q C L C R P M
Max score: -2
```cpp#include <iostream>#include <string>#include <vector>#include <algorithm>using namespace std;class Solution {const int MATCH = 1, MISMATCH = -1, GAP = -2;public:void printPath(int i, string &s, string &path1, int j, string &t, string &path2, vector<vector<int>> &dp, vector<vector<string>> &res) {// 使用回溯法输出所有最佳路径if (0 == i * j) {string tmp1 = path1 + string (i, '-');string tmp2 = path2 + string (i, '-');res.push_back({string (tmp1.rbegin(), tmp1.rend()),string (tmp2.rbegin(), tmp2.rend())});return;}// 在 t[j-1] 位置加入空位if (dp[i][j] == dp[i-1][j] + GAP) {// 选择path1.push_back(s[i-1]);path2.push_back('-');printPath(i - 1, s, path1, j, t, path2, dp, res);// 撤销path1.pop_back();path2.pop_back();}// 在 s[i-1] 位置加入空位if (dp[i][j] == dp[i][j-1] + GAP) {// 选择path1.push_back('-');path2.push_back(t[j-1]);printPath(i, s, path1, j - 1, t, path2, dp, res);// 撤销path1.pop_back();path2.pop_back();}// 不插入空位的情况if (dp[i][j] == dp[i-1][j-1] + (s[i-1] == t[j-1] ? MATCH: MISMATCH)) {// 选择path1.push_back(s[i-1]);path2.push_back(t[j-1]);printPath(i - 1, s, path1, j - 1, t, path2, dp, res);// 撤销path1.pop_back();path2.pop_back();}}vector<vector<string>> NededlemanWunsch(string &s, string &t) {int m = s.size(), n = t.size();// 全局比对算法: 完全匹配得 1 分,错配得 -1 分,插入空位得 -2 分// 定义 dp[i][j] 表示 s[0,i] 和 t[0,j] 的最大得分vector<vector<int>> dp(m + 1, vector<int> (n + 1, 0)); // 初始化 m+1 行,n+1 列d都为 0// 1. 状态初始化:两条序列任一为空时,另一非空序列只能插入 GAP(当两者都为空串得分0)for (int i = 1; i <= m; ++i) { // 当 t 为空dp[i][0] = dp[i-1][0] + 1;}for (int j = 1; j <= n; ++j) {dp[0][j] = dp[0][j-1] + 1;}// 2. 状态转移方程:// (1) 当前 s[i-1] != t[j-1] 时:此时要么其中一个插入 GAP,要么 s[i-1] 和 t[j-1] 错配// s 中插入空位(等价删除 t[j-1] 字符):问题转换为 求解 s[0,i-1] 和 t[0,j-2] 的最大的得分 + GAP// t 中插入空位(等价删除 s[i-1] 字符):问题转换为 求解 s[0,i-2] 和 t[0,j-1] 的最大的得分 + GAP// s[i-1] 和 t[j-1] 错配(强行对位):问题转换为 求解 s[0,i-2] 和 t[0,j-2] 的最大的得分 + MISMATCH// (2) 当前 s[i-1] = t[j-1] 时:即使当前匹配,但是也可能插入空位,牺牲当前匹配获得全局最优// s[i-1] 和 t[j-1] 匹配:问题转换为 求解 s[0,i-2] 和 t[0,j-2] 的最大的得分 + MATCH// s 中插入空位(等价删除 t[j-1] 字符):问题转换为 求解 s[0,i-1] 和 t[0,j-2] 的最大的得分 + GAP// t 中插入空位(等价删除 s[i-1] 字符):问题转换为 求解 s[0,i-2] 和 t[0,j-1] 的最大的得分 + GAPfor (int i = 1; i <= m; ++i) {for (int j = 1; j <= n; ++j) {dp[i][j] = max({dp[i-1][j-1] + (s[i-1] == t[j-1] ? MATCH: MISMATCH),dp[i-1][j] + GAP, // 在 t[j-1] 位置加入空位dp[i][j-1] + GAP // 在 s[i-1] 位置加入空位});}}cout << dp[m][n] << endl;// return dp[m][n];// 路径输出vector<vector<string>> res;string path1, path2;printPath(m, s, path1, n, t, path2, dp, res);return res;}};int main() {string seq1 = "ABCNJRQCLCRPM";string seq2 = "AJCJNRCKCRBP";vector<vector<string>> ret = Solution().NededlemanWunsch(seq1, seq2);for (const vector<string> & pathes: ret) {cout << pathes[0] << endl;cout << pathes[1] << endl;cout << endl;}return 0;}
:::tips
日常情况下,是不太要求输出全部结果的,因为这可能涉及到很大内存开销(几乎有  ),一般都不会出现这种来自两个方向甚至三个方向的可能!具体原因见下面分析!
:::
),一般都不会出现这种来自两个方向甚至三个方向的可能!具体原因见下面分析!
:::
6. 加入 BLOSUM62 矩阵
我们现在回归到生产应用中,理论结合实际。不再只是简单解决两个字符串问题,而是解决具有生物意义的问题。
除了两个氨基酸完全相同外,因为实际中用的是 BLOSUM62 替换矩阵,出现一定的相似性,也就是不匹配的时候(不加入空位)还可能有较多的分值。
- 单点(
.):在两序列中表示相似性弱;在序列边表示空位。 - 双点(
:):表示氨基酸有一定程度的相似性(比单点大,但比完全匹配小)
矩阵信息来自 NCBI
# Matrix made by matblas from blosum62.iij# * column uses minimum score# BLOSUM Clustered Scoring Matrix in 1/2 Bit Units# Blocks Database = /data/blocks_5.0/blocks.dat# Cluster Percentage: >= 62# Entropy = 0.6979, Expected = -0.5209A R N D C Q E G H I L K M F P S T W Y V B Z X *A 4 -1 -2 -2 0 -1 -1 0 -2 -1 -1 -1 -1 -2 -1 1 0 -3 -2 0 -2 -1 0 -4R -1 5 0 -2 -3 1 0 -2 0 -3 -2 2 -1 -3 -2 -1 -1 -3 -2 -3 -1 0 -1 -4N -2 0 6 1 -3 0 0 0 1 -3 -3 0 -2 -3 -2 1 0 -4 -2 -3 3 0 -1 -4D -2 -2 1 6 -3 0 2 -1 -1 -3 -4 -1 -3 -3 -1 0 -1 -4 -3 -3 4 1 -1 -4C 0 -3 -3 -3 9 -3 -4 -3 -3 -1 -1 -3 -1 -2 -3 -1 -1 -2 -2 -1 -3 -3 -2 -4Q -1 1 0 0 -3 5 2 -2 0 -3 -2 1 0 -3 -1 0 -1 -2 -1 -2 0 3 -1 -4E -1 0 0 2 -4 2 5 -2 0 -3 -3 1 -2 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4G 0 -2 0 -1 -3 -2 -2 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 -1 -2 -1 -4H -2 0 1 -1 -3 0 0 -2 8 -3 -3 -1 -2 -1 -2 -1 -2 -2 2 -3 0 0 -1 -4I -1 -3 -3 -3 -1 -3 -3 -4 -3 4 2 -3 1 0 -3 -2 -1 -3 -1 3 -3 -3 -1 -4L -1 -2 -3 -4 -1 -2 -3 -4 -3 2 4 -2 2 0 -3 -2 -1 -2 -1 1 -4 -3 -1 -4K -1 2 0 -1 -3 1 1 -2 -1 -3 -2 5 -1 -3 -1 0 -1 -3 -2 -2 0 1 -1 -4M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 0 -2 -1 -1 -1 -1 1 -3 -1 -1 -4F -2 -3 -3 -3 -2 -3 -3 -3 -1 0 0 -3 0 6 -4 -2 -2 1 3 -1 -3 -3 -1 -4P -1 -2 -2 -1 -3 -1 -1 -2 -2 -3 -3 -1 -2 -4 7 -1 -1 -4 -3 -2 -2 -1 -2 -4S 1 -1 1 0 -1 0 0 0 -1 -2 -2 0 -1 -2 -1 4 1 -3 -2 -2 0 0 0 -4T 0 -1 0 -1 -1 -1 -1 -2 -2 -1 -1 -1 -1 -2 -1 1 5 -2 -2 0 -1 -1 0 -4W -3 -3 -4 -4 -2 -2 -3 -2 -2 -3 -2 -3 -1 1 -4 -3 -2 11 2 -3 -4 -3 -2 -4Y -2 -2 -2 -3 -2 -1 -2 -3 2 -1 -1 -2 -1 3 -3 -2 -2 2 7 -1 -3 -2 -1 -4V 0 -3 -3 -3 -1 -2 -2 -3 -3 3 1 -2 1 -1 -2 -2 0 -3 -1 4 -3 -2 -1 -4B -2 -1 3 4 -3 0 1 -1 0 -3 -4 0 -3 -3 -2 0 -1 -4 -3 -3 4 1 -1 -4Z -1 0 0 1 -3 3 4 -2 0 -3 -3 1 -1 -3 -1 0 -1 -3 -2 -2 1 4 -1 -4X 0 -1 -1 -1 -2 -1 -1 -1 -1 -1 -1 -1 -1 -1 -2 0 0 -2 -1 -1 -1 -1 -1 -4* -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 1
其实实际上,现在替换矩阵和打分关系远比上面扣分标准变动很大。比如延长两个相邻空位不是扣 -2 分,还有更大的惩罚分。因此想要深入了解整个设计建议阅读源码等。基本概念和操作过程如上所述。
教科书上使用的 EMBOSS 程序中 needle 主页介绍在此,其在线打分程序点击此处
我们将上面的两条序列输入,打分参数选择默认,最终得到结果如下:
######################################### Program: needle# Rundate: Tue 10 Nov 2020 12:53:00# Commandline: needle# -auto# -stdout# -asequence emboss_needle-I20201110-125257-0884-69750982-p2m.asequence# -bsequence emboss_needle-I20201110-125257-0884-69750982-p2m.bsequence# -datafile EBLOSUM62# -gapopen 10.0# -gapextend 0.5# -endopen 10.0# -endextend 0.5# -aformat3 pair# -sprotein1# -sprotein2# Align_format: pair# Report_file: stdout#########################################=======================================## Aligned_sequences: 2# 1: EMBOSS_001# 2: EMBOSS_001# Matrix: EBLOSUM62# Gap_penalty: 10.0# Extend_penalty: 0.5## Length: 14# Identity: 7/14 (50.0%)# Similarity: 7/14 (50.0%)# Gaps: 3/14 (21.4%)# Score: 26.0###=======================================EMBOSS_001 1 ABCNJRQCLCR-PM 13|.|. | |.|| |EMBOSS_001 1 AJCJNR-CKCRBP- 12#---------------------------------------#---------------------------------------
可以看到,它的结果与我们按理想条件下得到的有较大的差距。

若有收获,就点个赞吧
0 人点赞
