- 基础题
- 1)统计reads_1.fq 文件中共有多少条序列信息
- 2)输出所有的reads_1.fq文件中的标识符(即以@开头的那一行)
- 3) 输出reads_1.fq文件中的 所有序列信息(即每个序列的第二行)
- 4)输出以‘@’及其后面的描述信息(即每个序列的第一行)
- 5)输出质量值信息(即每个序列的第四行)
- 6) 计算reads_1.fq 文件含有N碱基的reads个数
- 7) 统计文件中reads_1.fq文件里面的序列的碱基总数
- 8)计算reads_1.fq 所有的reads中
N碱基的总数 - 9)统计reads_1.fq 中测序碱基质量值恰好为
Q20的个数 - 10)统计reads_1.fq 中测序碱基质量值恰好为
Q30的个数 - 11)统计reads_1.fq 中所有序列的第一位
碱基的ATCGNatcg分布情况 - 12)将reads_1.fq 转为reads_1.fa文件(即将fastq转化为fasta)
- 13) 统计上述reads_1.fa文件中共有多少条序列
- 14)计算reads_1.fa文件中总的碱基序列的GC数量
- 15)删除 reads_1.fa文件中的每条序列的N碱基
- 16)删除 reads_1.fa文件中的含有N碱基的序列
- 17) 删除 reads_1.fa文件中的短于65bp的序列
- 18) 删除 reads_1.fa文件每条序列的前后五个碱基
- 19)删除 reads_1.fa文件中的长于125bp的序列
- 20)查看reads_1.fq 中每条序列的第一位碱基的质量值的平均值
基础题
如果涉及到一些没讲过的知识点,可以自己尝试搜索一下,实在不会做就跳过,能做多少就多少
1)统计reads_1.fq 文件中共有多少条序列信息
首先要了解一下fq文件的数据格式,每4行为一条序列的信息
1.序列标识以及相关的描述信息,以‘@’开头;
2.是碱基序列
3.以‘+’开头,一般后面什么都不加,或者加序列标示符、描述信息,为预留行
4.是质量信息,和第二行的序列相对应,每一个序列都有一个质量评分,根据评分体系的不同,每个字符的含义表示的数字也不相同。
$ cat reads_1.fq | grep '^@r' | wc -l10000
2)输出所有的reads_1.fq文件中的标识符(即以@开头的那一行)
$ cat reads_1.fq | grep '^@r' | less
3) 输出reads_1.fq文件中的 所有序列信息(即每个序列的第二行)
$ cat reads_1.fq | sed -n '2~4p' | less
4)输出以‘@’及其后面的描述信息(即每个序列的第一行)
$ cat reads_1.fq | sed -n '1~4p' | less
5)输出质量值信息(即每个序列的第四行)
$ cat reads_1.fq | sed -n '4~4p' | less
6) 计算reads_1.fq 文件含有N碱基的reads个数
$ cat reads_1.fq | sed -n '2~4p' | grep 'N' | wc -l
6429
$ cat reads_1.fq | sed -n '2~4p' | grep -c 'N'
6429
7) 统计文件中reads_1.fq文件里面的序列的碱基总数
$ $ cat -A reads_1.fq | sed -n '2~4p' | wc
10000 10000 1108399
$ echo "1108399-10000" | bc
1098399
8)计算reads_1.fq 所有的reads中N碱基的总数
cat reads_1.fq | sed -n '2~4p' | grep -o 'N' | wc -l
26001
9)统计reads_1.fq 中测序碱基质量值恰好为Q20的个数
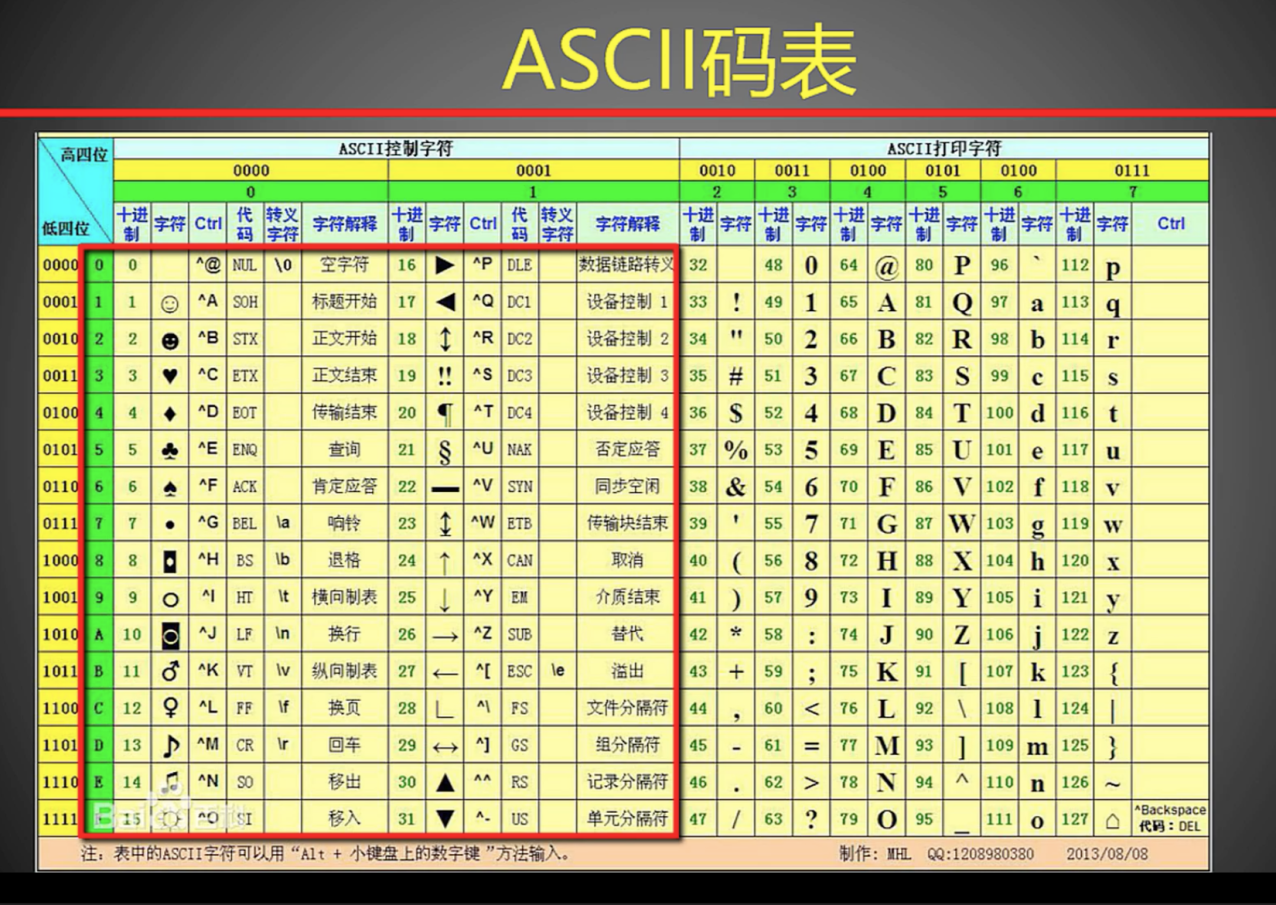
判断出是 Phred33 体系的质量值,因此Q20对应的ASCII码为 5 。
$ cat reads_1.fq | sed -n '4~4p' | grep -o '5' | wc -l
21369
10)统计reads_1.fq 中测序碱基质量值恰好为Q30的个数
$ cat reads_1.fq | sed -n '4~4p' | grep -o '?' | wc -l
21574
11)统计reads_1.fq 中所有序列的第一位碱基的ATCGNatcg分布情况
$ cat reads_1.fq | sed -n '2~4p' | sed -n '/^A/p' | wc -l
2184
Aug15 15:35:13 ~/work/Data
$ cat reads_1.fq | sed -n '2~4p' | sed -n '/^T/p' | wc -l
2253
Aug15 15:35:38 ~/work/Data
$ cat reads_1.fq | sed -n '2~4p' | sed -n '/^C/p' | wc -l
2203
Aug15 15:35:42 ~/work/Data
$ cat reads_1.fq | sed -n '2~4p' | sed -n '/^G/p' | wc -l
2219
Aug15 15:35:44 ~/work/Data
$ cat reads_1.fq | sed -n '2~4p' | sed -n '/^N/p' | wc -l
1141
12)将reads_1.fq 转为reads_1.fa文件(即将fastq转化为fasta)
$ cat reads_1.fq | sed -ne '1~4p' -ne '2~4p' | tr '@' '>' > reads_1.fa
13) 统计上述reads_1.fa文件中共有多少条序列
$ grep '>' reads_1.fa | wc -l
10000
14)计算reads_1.fa文件中总的碱基序列的GC数量
$ cat reads_1.fa | sed -n '2~2p'|grep -o -e 'CG' -o -e 'GC'|wc -l
113624
15)删除 reads_1.fa文件中的每条序列的N碱基
$ cat reads_1.fa | tr -d 'N'
16)删除 reads_1.fa文件中的含有N碱基的序列
17) 删除 reads_1.fa文件中的短于65bp的序列
18) 删除 reads_1.fa文件每条序列的前后五个碱基
19)删除 reads_1.fa文件中的长于125bp的序列
20)查看reads_1.fq 中每条序列的第一位碱基的质量值的平均值

若有收获,就点个赞吧
0 人点赞
