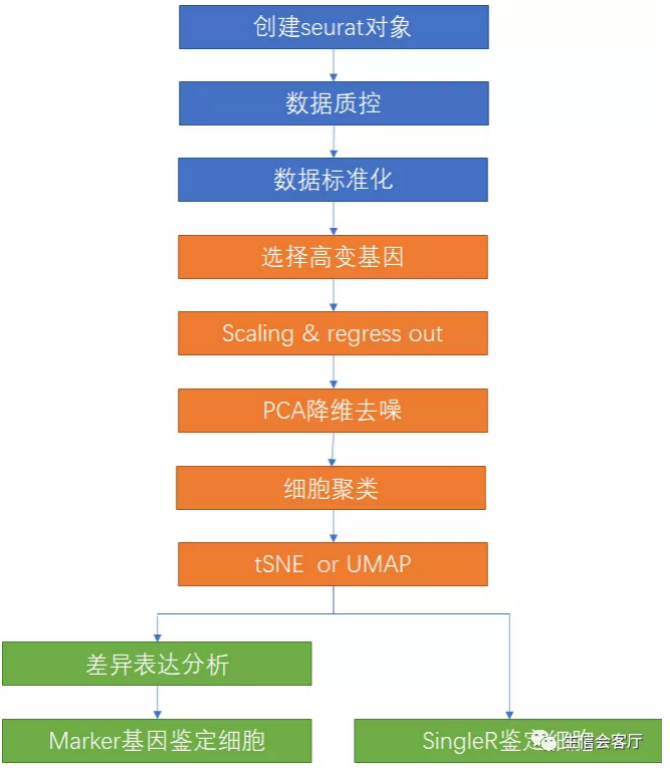
rm(list=ls())library(dplyr)library(Seurat)library(patchwork)## =============1.Load the PBMC datasetdata_dir <- "data/pbmc3k_filtered_gene_bc_matrices/"pbmc.data <- Read10X(data.dir = data_dir)pbmc.data# Initialize the Seurat object with the raw (non-normalized data)# min.cell:每个feature至少在多少个细胞中表达# min.features:每个细胞中至少有多少个feature被检测到pbmc <- CreateSeuratObject(counts = pbmc.data,project = "pbmc3k",min.cells = 3,min.features = 200)pbmc## =============2.探索一下数据# count matrix长什么样?# 首先看看三个基因的count值,'.'values in the matrix represent 0s (no molecules detected)pbmc.data[1:30, 1:30]pbmc.data[c("CD3D", "TCL1A", "MS4A1"), 1:30]rownames(pbmc.data)colnames(pbmc.data)# 0值矩阵大小raw_counts <- as.matrix(pbmc.data)raw_counts[1:4,1:4]dense.size <- object.size(raw_counts)dense.size# 稀疏矩阵大小sparse.size <- object.size(pbmc.data)sparse.size# 压缩比dense.size/sparse.size## =============3.QC:# 1.每个细胞中RNA的个数;# 2,每个细胞中线粒体基因表达的比例# The [[ operator can add columns to object metadata. This is a great place to stash QC statsgrep(pattern = "^MT-", rownames(pbmc), value = T)pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")# QC指标head(pbmc@meta.data, 5)# QC指标使用小提琴图可视化VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"),ncol = 3,pt.size = 0.1)# 两个指标之间的相关性plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA",feature2 = "percent.mt")plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA",feature2 = "nFeature_RNA")plot1 + plot2# 过滤pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)pbmc## =============4.标准化# LogNormalizepbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize",scale.factor = 10000)# 标准化后的值保存在:pbmc[["RNA"]]@datanormalized.data <- pbmc[["RNA"]]@datanormalized.data[1:20,1:4]dim(normalized.data)## =============5.鉴定高变基因# 高变基因:在一些细胞中表达高,另一些细胞中表达低的基因# 变异指标: mean-variance relationship# 默认返回两千个高变基因,用于下游如PCA降维分析。pbmc <- FindVariableFeatures(pbmc, selection.method = "vst",nfeatures = 2000)# 提取前10的高变基因top10 <- head(VariableFeatures(pbmc), 10)top10# 展示高变基因plot1 <- VariableFeaturePlot(pbmc)plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)plot1plot2## =============6.Scaling the data# 归一化处理:每一个基因在所有细胞中的均值变为0,方差标为1,对于降维来说是必需步骤# 归一化后的值保存在:pbmc[["RNA"]]@scale.datapbmc <- ScaleData(pbmc)scale.data <- pbmc[["RNA"]]@scale.datadim(scale.data)scale.data[1:10,1:4]# 后面绘制热图使用的是归一化后的值,可以选择全部基因归一化,但是耗时久一点all.genes <- rownames(pbmc)pbmc <- ScaleData(pbmc, features = all.genes)## =============7.降维# PCA降维,默认使用前面2000个高变基因,可以使用features改变用于降维的基因集pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc))# 可视化VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")DimPlot(pbmc, reduction = "pca")DimHeatmap(pbmc, dims = 1, cells = 500, balanced = TRUE)DimHeatmap(pbmc, dims = 1:15, cells = 500, balanced = TRUE)## =============8.确定使用PC个数# # each PC essentially representing a ‘metafeature’# 方式1:根据p值判断,比较耗时# pbmc <- JackStraw(pbmc, num.replicate = 100)# pbmc <- ScoreJackStraw(pbmc, dims = 1:20)## JackStrawPlot(pbmc, dims = 1:15)# 方式2:肘部法,根据拐点判断,比较主观,尽量多选几个PCElbowPlot(pbmc, ndims = 30)## =============9.对细胞聚类# 选取10个PC,首先基于PCA空间构建一个基于欧氏距离的KNN图pbmc <- FindNeighbors(pbmc, dims = 1:10)# 聚类并最优化# resolution参数:值越大,细胞分群数越多,# 0.4-1.2 typically returns good results for single-cell datasets of around 3K cells# Optimal resolution often increases for larger datasets.pbmc <- FindClusters(pbmc, resolution = 0.5)# 查看聚类数IDhead(Idents(pbmc), 5)# 查看每个类别多少个细胞head(pbmc@meta.data)table(pbmc@meta.data$seurat_clusters)# resolution对聚类的影响res.used <- seq(0.1,1,by=0.2)res.usedfor(i in res.used){pbmc <- FindClusters(pbmc, verbose = T, resolution = res.used)}# 可视化library(clustree)clus.tree.out <- clustree(pbmc) +theme(legend.position = "bottom") +scale_color_brewer(palette = "Set1") +scale_edge_color_continuous(low = "grey80", high = "red")clus.tree.outpbmc <- FindClusters(pbmc, verbose = T, resolution = 0.5)## =============10.将细胞在低维空间可视化UMAP/tSNEset.seed(123)pbmc <- RunUMAP(pbmc, dims = 1:10)pbmc <- RunTSNE(pbmc, dims = 1:10)# 可视化DimPlot(pbmc, reduction = "umap", label = T, label.size = 5)DimPlot(pbmc, reduction = "tsne", label = T, label.size = 5)saveRDS(pbmc, file = "data/pbmc_tutorial.rds")#pbmc <- readRDS("data/pbmc_tutorial.rds")## =============11.差异表达分析# 在cluster2 vs else中差异表达cluster1.markers <- FindMarkers(pbmc, ident.1 = 2, min.pct = 0.25)head(cluster1.markers, n = 5)# 指定两个类cluster 5 from clusters 0 and 3cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25)head(cluster5.markers, n = 5)# 所有类的差异表达基因# only.pos:只保留上调差异表达的基因pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)write.csv(pbmc.markers,file = "data/pbmc.markers.csv")head(pbmc.markers)# 筛选pbmc.markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_log2FC)# 选择一些基因进行可视化,作者这里根据自己的知识背景选择的相关基因VlnPlot(pbmc, features = c("MS4A1", "CD79A"))VlnPlot(pbmc, features = c("NKG7", "PF4"), slot = "counts", log = TRUE)FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A"))DotPlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A")) + coord_flip() +theme(axis.text.x = element_text(angle=90, hjust=1, vjust=.5))top10 <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_log2FC)DoHeatmap(pbmc, features = top10$gene) + NoLegend()## =============12.细胞类型鉴定:使用经典的markers进行注释new.cluster.ids <- c("Naive CD4 T", # IL7R, CCR7"CD14+ Mono", # CD14, LYZ"Memory CD4 T", # IL7R, S100A4"B", # MS4A1"CD8 T", # CD8A"FCGR3A+ Mono", # FCGR3A, MS4A7"NK", # GNLY, NKG7"DC", # FCER1A, CST3"Platelet") # PPBPnames(new.cluster.ids) <- levels(pbmc)new.cluster.ids# 原来的细胞聚类名称Idents(pbmc)# 更改细胞聚类的名字pbmc <- RenameIdents(pbmc, new.cluster.ids)Idents(pbmc)# 可视化DimPlot(pbmc, reduction = "umap", label = TRUE, pt.size = 2,label.box = T) + NoLegend()DimPlot(pbmc, reduction = "tsne", label = TRUE, pt.size = 2,label.box = T) + NoLegend()# 保存结果pbmc@meta.data$cell_anno <- Idents(pbmc)head(pbmc@meta.data)write.csv(pbmc@meta.data,file = "data/metadata.csv")saveRDS(pbmc, file = "data/pbmc3k_final.rds")table(pbmc@meta.data$cell_anno)## 去掉画图部分的流程总结data_dir <- "data/pbmc3k_filtered_gene_bc_matrices/"pbmc.counts <- Read10X(data.dir = data_dir)pbmc <- CreateSeuratObject(counts = pbmc.counts)pbmc <- NormalizeData(object = pbmc)pbmc <- FindVariableFeatures(object = pbmc)pbmc <- ScaleData(object = pbmc)pbmc <- RunPCA(object = pbmc)pbmc <- FindNeighbors(object = pbmc)pbmc <- FindClusters(object = pbmc)pbmc <- RunTSNE(object = pbmc)pbmc <- RunUMAP(object = pbmc)DimPlot(object = pbmc, reduction = "tsne")
代码及图片均来自于生信技能树张娟老师

若有收获,就点个赞吧
0 人点赞
