一、fastQC
# 激活conda环境conda activate rna# 连接数据到自己的文件夹cd $HOME/project/Human-16-Asthma-Trans/data/rawdataln -s /home/t_rna/data/airway/fastq_raw25000/*gz ./# 使用FastQC软件对单个fastq文件进行质量评估,结果输出到qc/文件夹下nohup fastqc -t 6 -o ./ SRR*.fastq.gz >qc.log & ##后台挂起# 使用MultiQc整合FastQC结果# 使用绝对路径运行multiqc=/home/t_rna/miniconda3/envs/rna/bin/multiqcfastqc=/home/t_rna/miniconda3/envs/rna/bin/fastqcfq_dir=$HOME/project/Human-16-Asthma-Trans/data/rawdataoutdir=$HOME/project/Human-16-Asthma-Trans/data/rawdata# 使用绝对路径运行# $fastqc -t 6 -o $outdir ${fq_dir}/SRR*.fastq.gz >${fq_dir}/qc.log# 报告整合$multiqc $outdir/*.zip -o $outdir/ >${fq_dir}/multiqc.log

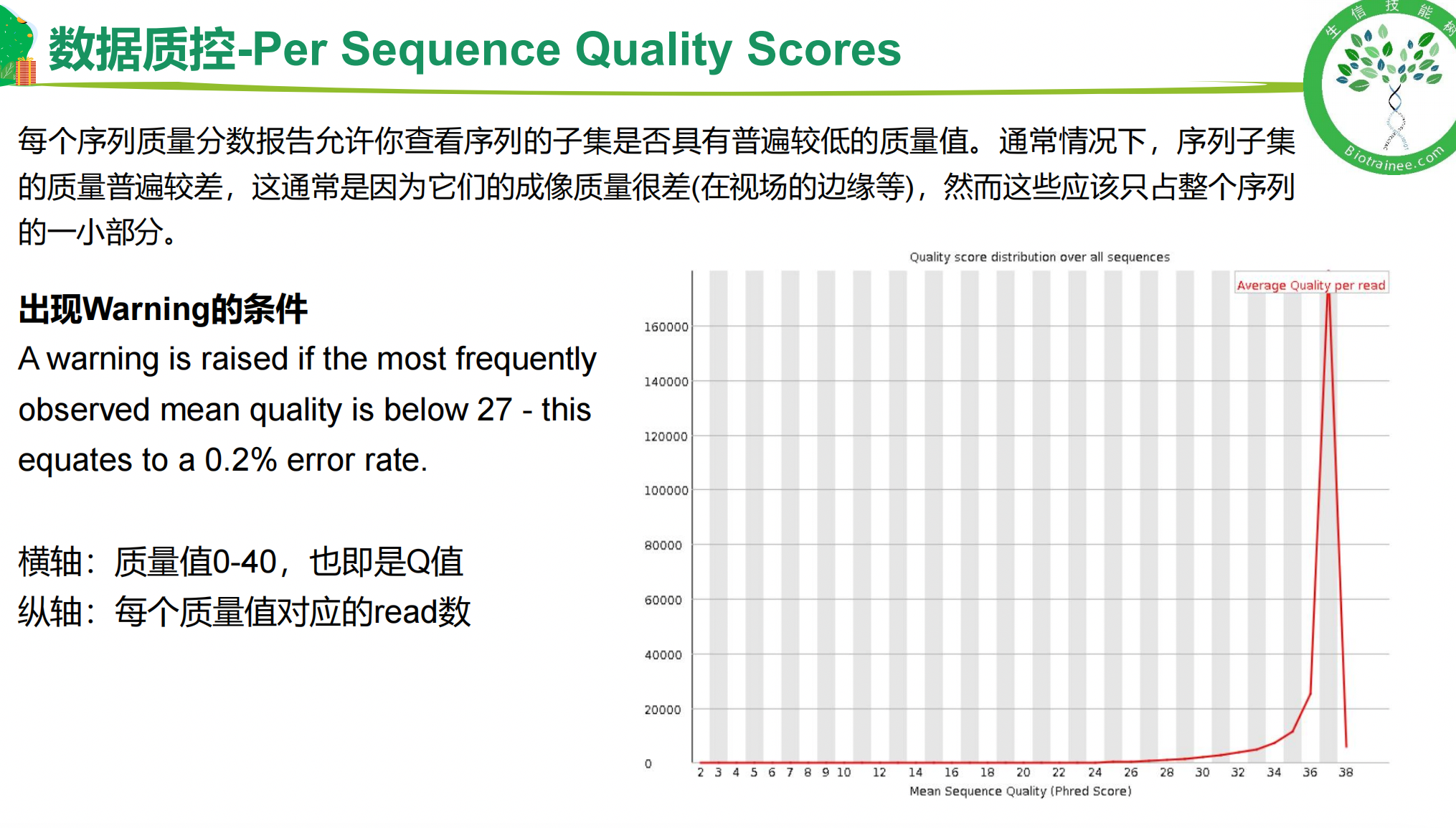
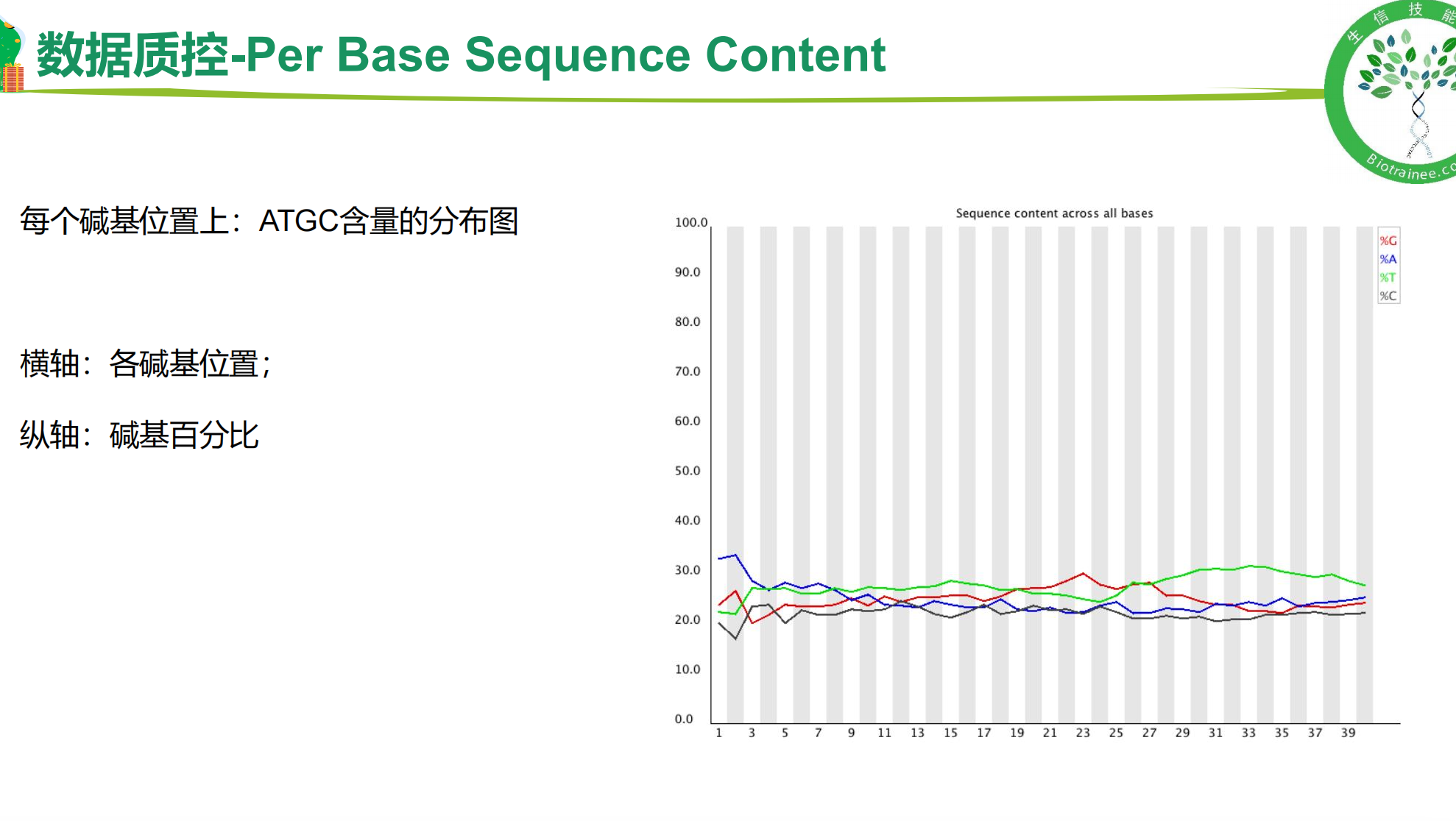
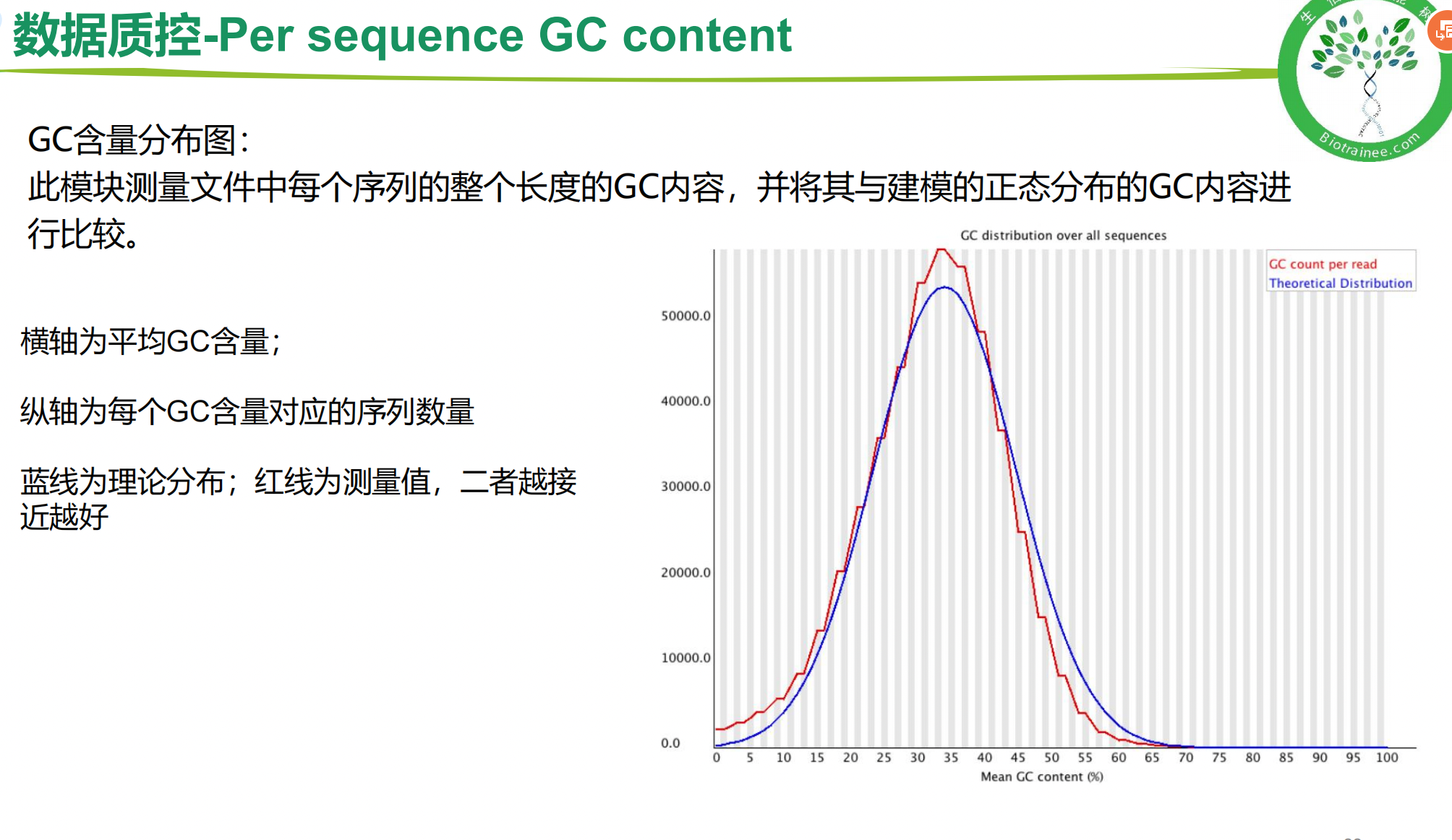
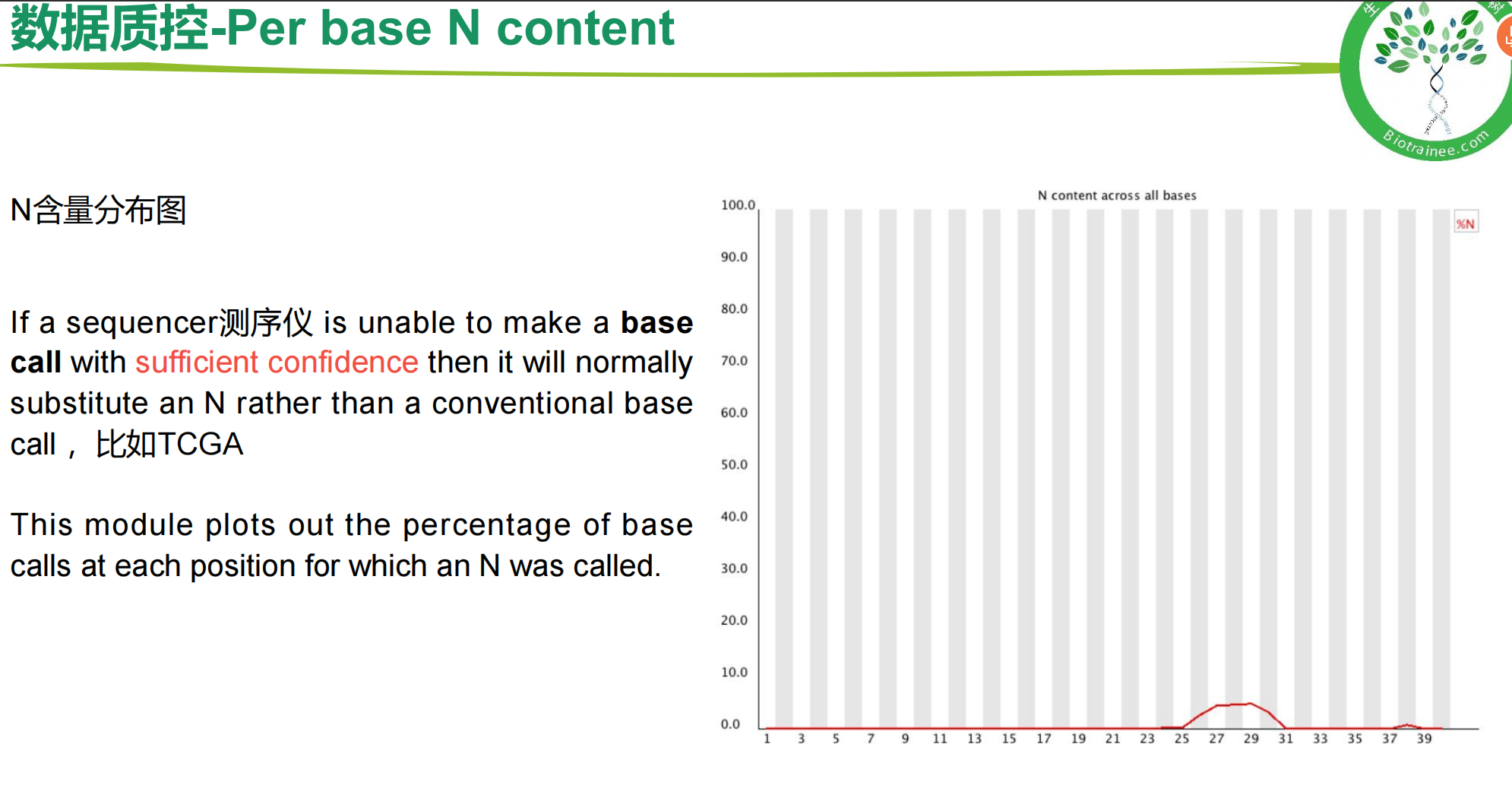
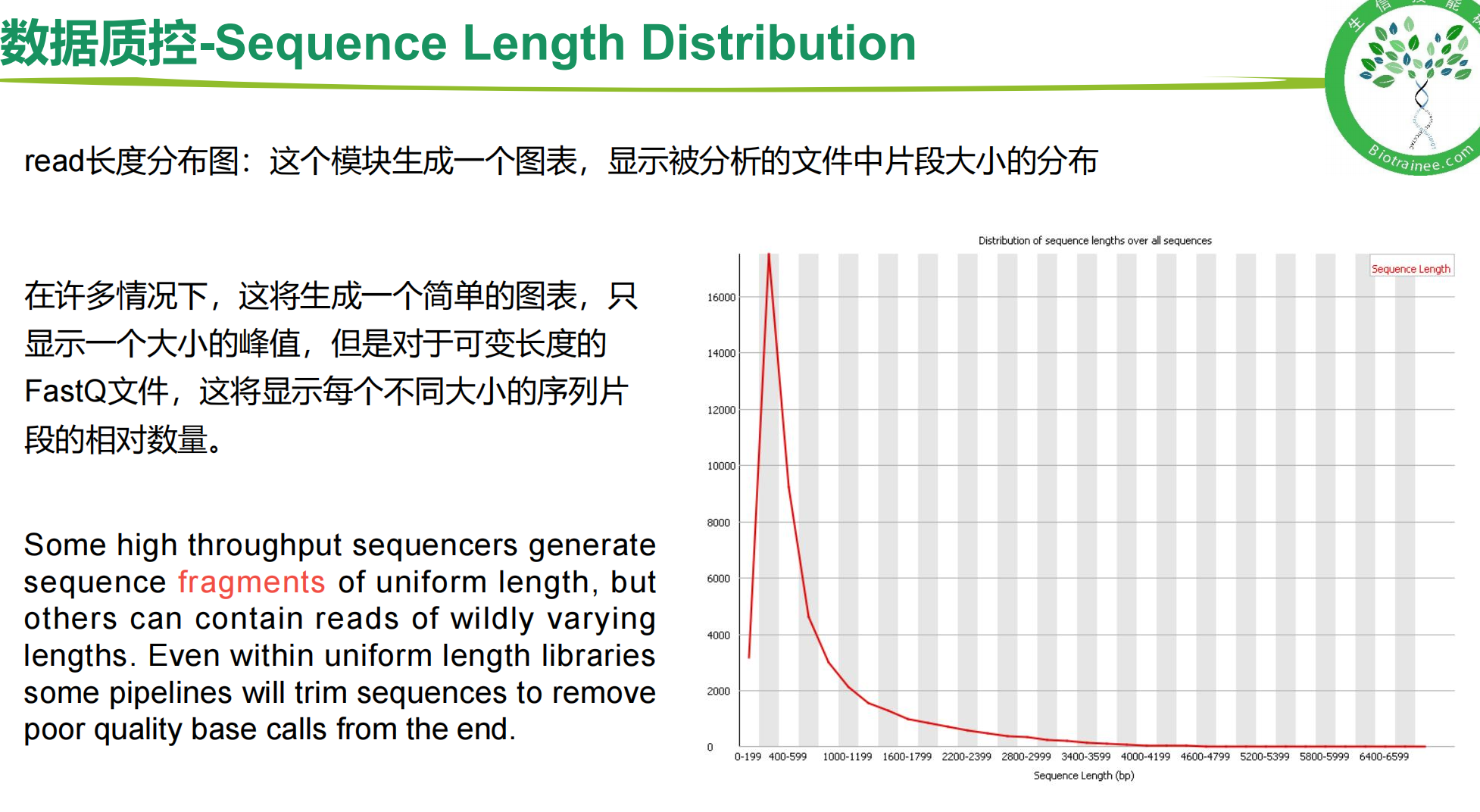
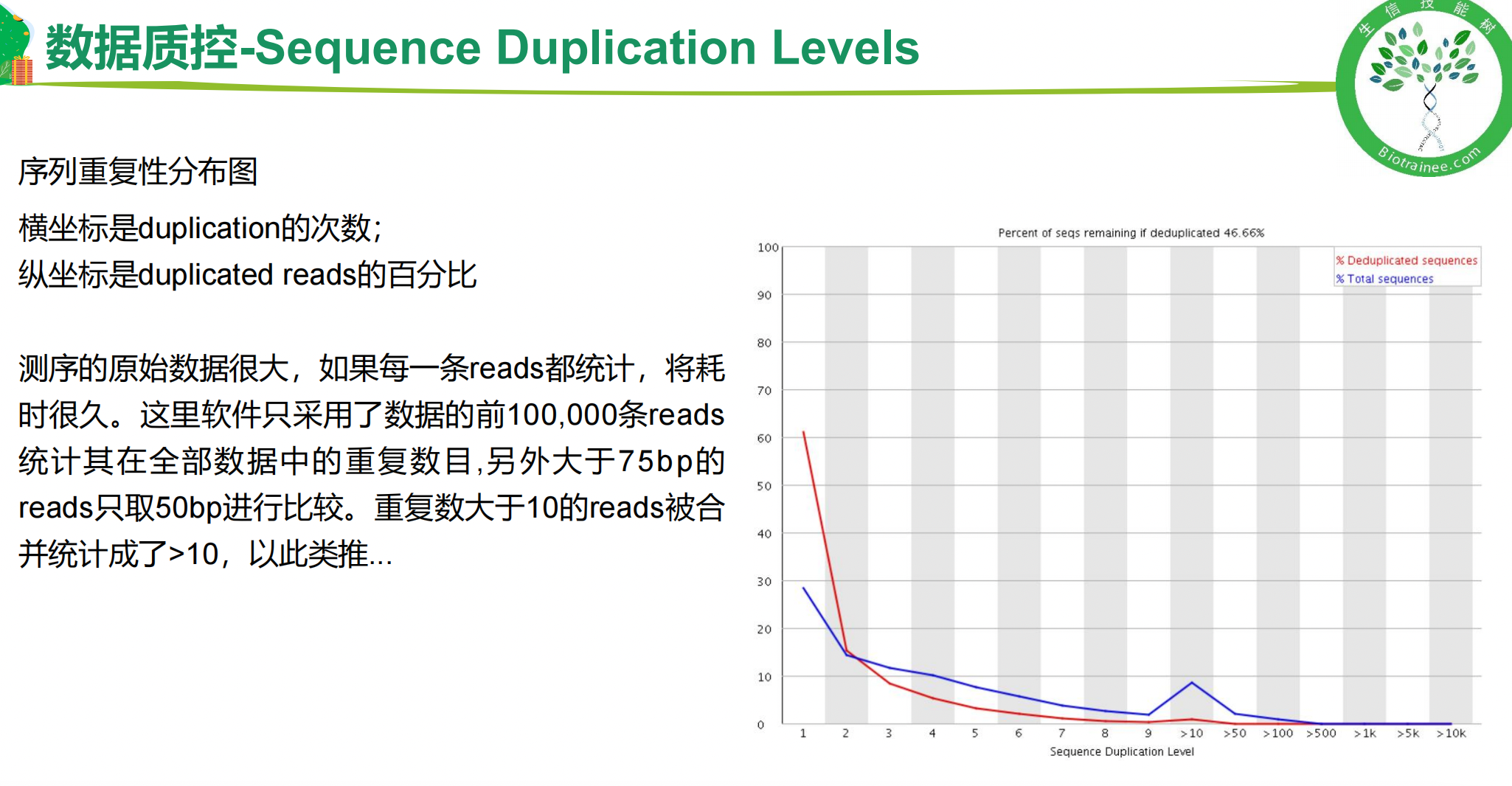

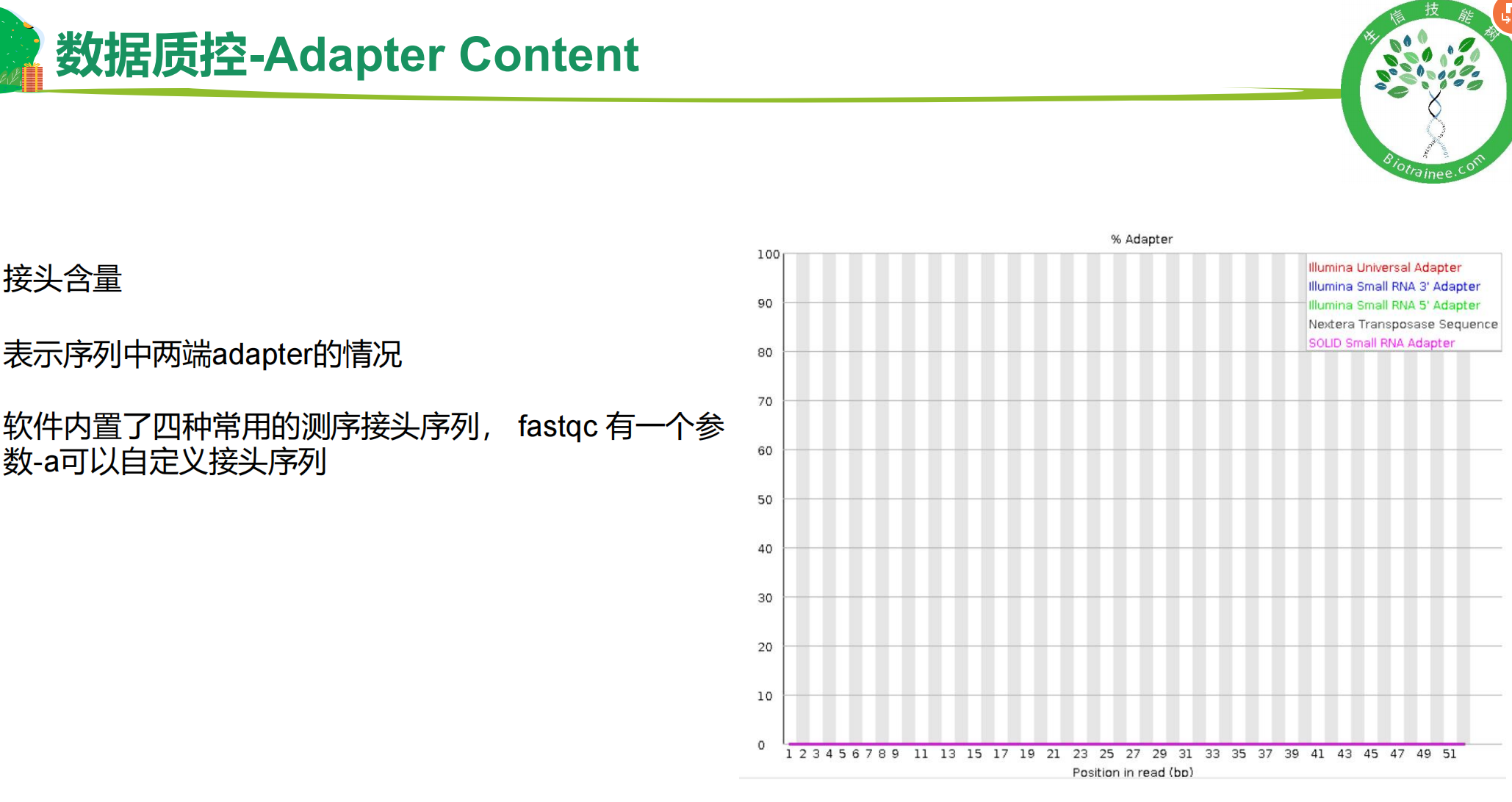
二、过滤
1、trim_galore
# 激活小环境
conda activate rna
# 新建文件夹trim_galore
cd $HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
# 先生成一个变量,为样本ID
ls $HOME/project/Human-16-Asthma-Trans/data/rawdata/*_1.fastq.gz | awk -F'/' '{print $NF}' | cut -d'_' -f1 >ID
# 多个样本 vim trim_galore.sh,以下为sh的内容
rawdata=$HOME/project/Human-16-Asthma-Trans/data/rawdata
cleandata=$HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
cat ID | while read id
do
trim_galore -q 20 --length 20 --max_n 3 --stringency 3 --fastqc --paired -o ${cleandata} ${rawdata}/${id}_1.fastq.gz ${rawdata}/${id}_2.fastq.gz
done
# 提交任务到后台
nohup sh trim_galore.sh >trim_galore.log &
# 使用MultiQc整合FastQC结果
multiqc *.zip
## ==============================================
## 补充技巧:使用掐头去尾获得样本ID
ls $rawdata/*_1.fastq.gz | while read id
do
name=${id##*/}
name=${name%_*}
echo "trim_galore -q 20 --length 20 --max_n 3 --stringency 3 --fastqc --paired -o ${cleandata} ${rawdata}/${name}_1.fastq.gz ${rawdata}/${name}_2.fastq.gz "
done
2、fastp
cd $HOME/project/Human-16-Asthma-Trans/data/cleandata/fastp
# 定义文件夹:vim fastp.sh
cleandata=$HOME/project/Human-16-Asthma-Trans/data/cleandata/fastp/
rawdata=$HOME/project/Human-16-Asthma-Trans/data/rawdata/
cat ../trim_galore/ID | while read id
do
fastp -l 20 -q 20 --compression=6 \
-i ${rawdata}/${id}_1.fastq.gz \
-I ${rawdata}/${id}_2.fastq.gz \
-o ${cleandata}/${id}_clean_1.fq.gz \
-O ${cleandata}/${id}_clean_2.fq.gz \
-R ${cleandata}/${id} \
-h ${cleandata}/${id}.fastp.html \
-j ${cleandata}/${id}.fastp.json
done
# 运行fastp脚本
nohup sh fastp.sh >fastp.log &
3、数据过滤前后的比较
# 进入过滤目录
cd $HOME/project/Human-16-Asthma-Trans/data/cleandata/trim_galore
# 原始数据
zcat $rawdata/SRR1039510_1.fastq.gz | paste - - - - > raw.txt
# 过滤后的数据
zcat SRR1039510_1_val_1.fq.gz |paste - - - - > trim.txt
awk '(length($4)<63){print$1}' trim.txt > Seq.ID
head -n 100 Seq.ID > ID100
grep -w -f ID100 trim.txt | awk '{print$1,$4}' > trim.sm
grep -w -f ID100 raw.txt | awk '{print$1,$4}' > raw.sm
paste raw.sm trim.sm | awk '{print$2,$4}' | tr ' ' '\n' |less -S
代码及图片均来自于生信技能树张娟老师

若有收获,就点个赞吧
0 人点赞
