Find Variation Using Pilon
The input genome is the NC_045512.2 ref genome and the BAM file is a subset of alignment results with ref genome.
java -Xmx64G -jar ~/miniconda3/envs/covid/share/pilon-1.23-3/pilon-1.23.jar \--genome ~/SARS_CoV_2/mapping/ref/Wuhan-Hu-1.fasta \--frags ~/SARS_CoV_2/subsample/P3-VERO-P3-${i}-vero_L4_sort_0.1.bam \--threads 32 \--changes \--output ${i}.fasta \--fix all,breaks\--vcf &> pilon${i}.log
· View .changes File
The .changes file contains a space-delimited record of every change made in the assembly as instructed by the --fix option.
for i in {1..11}; do{echo ">${i}.fasta.changes" >> sum.txtcat ${i}.fasta.changes >>sum.txt}done
>1.fasta.changesNC_045512.2:11083 NC_045512.2_pilon:11083 G TNC_045512.2:23607 NC_045512.2_pilon:23607 G ANC_045512.2:26144 NC_045512.2_pilon:26144 G T>2.fasta.changesNC_045512.2:8782 NC_045512.2_pilon:8782 C TNC_045512.2:28144 NC_045512.2_pilon:28144 T C>3.fasta.changesNC_045512.2:16846 NC_045512.2_pilon:16846 G T>4.fasta.changes>5.fasta.changes>6.fasta.changesNC_045512.2:3885 NC_045512.2_pilon:3885 C ANC_045512.2:12778 NC_045512.2_pilon:12778 C TNC_045512.2:13270 NC_045512.2_pilon:13270 T CNC_045512.2:29449 NC_045512.2_pilon:29449 G T>7.fasta.changesNC_045512.2:1405 NC_045512.2_pilon:1405 T CNC_045512.2:9802 NC_045512.2_pilon:9802 G TNC_045512.2:19945 NC_045512.2_pilon:19945 A GNC_045512.2:25267 NC_045512.2_pilon:25267 C TNC_045512.2:27615 NC_045512.2_pilon:27615 C G>8.fasta.changesNC_045512.2:8782 NC_045512.2_pilon:8782 C TNC_045512.2:18060 NC_045512.2_pilon:18060 C TNC_045512.2:28144 NC_045512.2_pilon:28144 T C>9.fasta.changesNC_045512.2:22303 NC_045512.2_pilon:22303 T G>10.fasta.changesNC_045512.2:16114 NC_045512.2_pilon:16114 C TNC_045512.2:22205 NC_045512.2_pilon:22205 G C>11.fasta.changesNC_045512.2:22303 NC_045512.2_pilon:22303 T GNC_045512.2:27775 NC_045512.2_pilon:27775 T CNC_045512.2:27776 NC_045512.2_pilon:27776 T GNC_045512.2:27777 NC_045512.2_pilon:27777 G A
· view .vcf file
The Variant Call Format (VCF) specifies the format of a text file used for storing gene sequence variations. It contains meta-information lines, a header line, and then data lines each containing information about a position in the genome.
Official document: https://samtools.github.io/hts-specs/VCFv4.2.pdf
Example:
$ cat 1.fasta.vcf | head -n 50##fileformat=VCFv4.1##fileDate=20210428##source="Pilon version 1.23 Mon Nov 26 16:04:05 2018 -0500"##PILON="--genome /public/home/ykk/SARS_CoV_2/Pilon/wuhan/Wuhan-Hu-1.fasta --frags /public/home/ykk/SARS_CoV_2/sortedbam/P3-VERO-P3-1-vero_L4_sort.bam --changes --output 1.fasta --threads 32 --fix all,breaks --vcf"##reference=file:/public/home/ykk/SARS_CoV_2/Pilon/wuhan/Wuhan-Hu-1.fasta##contig=<ID=NC_045512.2,length=29903>##FILTER=<ID=LowCov,Description="Low Coverage of good reads at location">##FILTER=<ID=Amb,Description="Ambiguous evidence in haploid genome">##FILTER=<ID=Del,Description="This base is in a deletion or change event from another record">##INFO=<ID=DP,Number=1,Type=Integer,Description="Valid read depth; some reads may have been filtered">##INFO=<ID=TD,Number=1,Type=Integer,Description="Total read depth including bad pairs">##INFO=<ID=PC,Number=1,Type=Integer,Description="Physical coverage of valid inserts across locus">##INFO=<ID=BQ,Number=1,Type=Integer,Description="Mean base quality at locus">##INFO=<ID=MQ,Number=1,Type=Integer,Description="Mean read mapping quality at locus">##INFO=<ID=QD,Number=1,Type=Integer,Description="Variant confidence/quality by depth">##INFO=<ID=BC,Number=4,Type=Integer,Description="Count of As, Cs, Gs, Ts at locus">##INFO=<ID=QP,Number=4,Type=Integer,Description="Percentage of As, Cs, Gs, Ts weighted by Q & MQ at locus">##INFO=<ID=IC,Number=1,Type=Integer,Description="Number of reads with insertion here">##INFO=<ID=DC,Number=1,Type=Integer,Description="Number of reads with deletion here">##INFO=<ID=XC,Number=1,Type=Integer,Description="Number of reads clipped here">##INFO=<ID=AC,Number=A,Type=Integer,Description="Allele count in genotypes, for each ALT allele, in the same order as listed">##INFO=<ID=AF,Number=A,Type=Float,Description="Fraction of evidence in support of alternate allele(s)">##INFO=<ID=SVTYPE,Number=1,Type=String,Description="Type of structural variant">##INFO=<ID=SVLEN,Number=.,Type=String,Description="Difference in length between REF and ALT alleles">##INFO=<ID=END,Number=1,Type=Integer,Description="End position of the variant described in this record">##INFO=<ID=IMPRECISE,Number=0,Type=Flag,Description="Imprecise change from local reassembly (ALT contains Ns)">##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">##FORMAT=<ID=AD,Number=.,Type=String,Description="Allelic depths for the ref and alt alleles in the order listed">##FORMAT=<ID=DP,Number=1,Type=String,Description="Approximate read depth; some reads may have been filtered">##ALT=<ID=DUP,Description="Possible segmental duplication">#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT SAMPLENC_045512.2 1 . A . 359 LowCov DP=10;TD=2439;BQ=36;MQ=34;QD=35;BC=10,0,0,0;QP=100,0,0,0;PC=11;IC=0;DC=0;XC=29;AC=0;AF=0.00 GT0/0NC_045512.2 2 . T . 370 LowCov DP=10;TD=2473;BQ=37;MQ=34;QD=37;BC=0,0,0,10;QP=0,0,0,100;PC=11;IC=0;DC=0;XC=57;AC=0;AF=0.00 GT0/0…………NC_045512.2 11083 . G T 11822391 PASS DP=453118;TD=466945;BQ=36;MQ=60;QD=30;BC=178,42,363,385402;QP=0,0,0,100;PC=410199;IC=10;DC=67133;XC=3036;AC=2;AF=1.00 GT1/1…………NC_045512.2 23607 . G A 78429249 PASS DP=2706046;TD=2821970;BQ=35;MQ=60;QD=28;BC=2582038,3758,118877,1272;QP=95,0,4,0;PC=2977892;IC=1;DC=101;XC=29688;AC=2;AF=0.83 GT1/1…………NC_045512.2 26144 . G T 56706266 PASS DP=1578759;TD=1610205;BQ=36;MQ=60;QD=35;BC=1041,234,2028,1575401;QP=0,0,0,100;PC=1396070;IC=82;DC=55;XC=3296;AC=2;AF=0.00 GT1/1…………
GT1/1 indicates that the site in the sample is a mutation, and mutation type is the same as the column ALT‘s base type. The 1.fasta.vcf file above shows that there are 3 mutations in the first genome, which is consistent with the content of the 1.fasta.changes file.
4. Visualize vcf file.
- Install
DISCVR-seq,Picardandgatk4toolkit.
git clone https://github.com/BimberLab/DISCVRSeq/git clone https://github.com/broadinstitute/picard.gitconda install gatk4 -y
Build index for ref genome.
samtools faidx Wuhan-Hu-1.fastajava -jar ~/software/picard.jar CreateSequenceDictionary R=Wuhan-Hu-1.fasta O=Wuhan-Hu-1.dict
Build index for
vcffile usingGATK4.java -jar ~/miniconda3/envs/covid/share/gatk4-4.2.0.0-1/gatk-package-4.2.0.0-local.jar IndexFeatureFile -I ~/SARS_CoV_2/Pilon/1.fasta.vcf
Visualize
vcffile.java -jar ~/software/DISCVRSeq-1.26.jar VariantQC -O 1.html -R Wuhan-Hu-1.fasta -V ~/SARS_CoV_2/Pilon/1.fasta.vcf
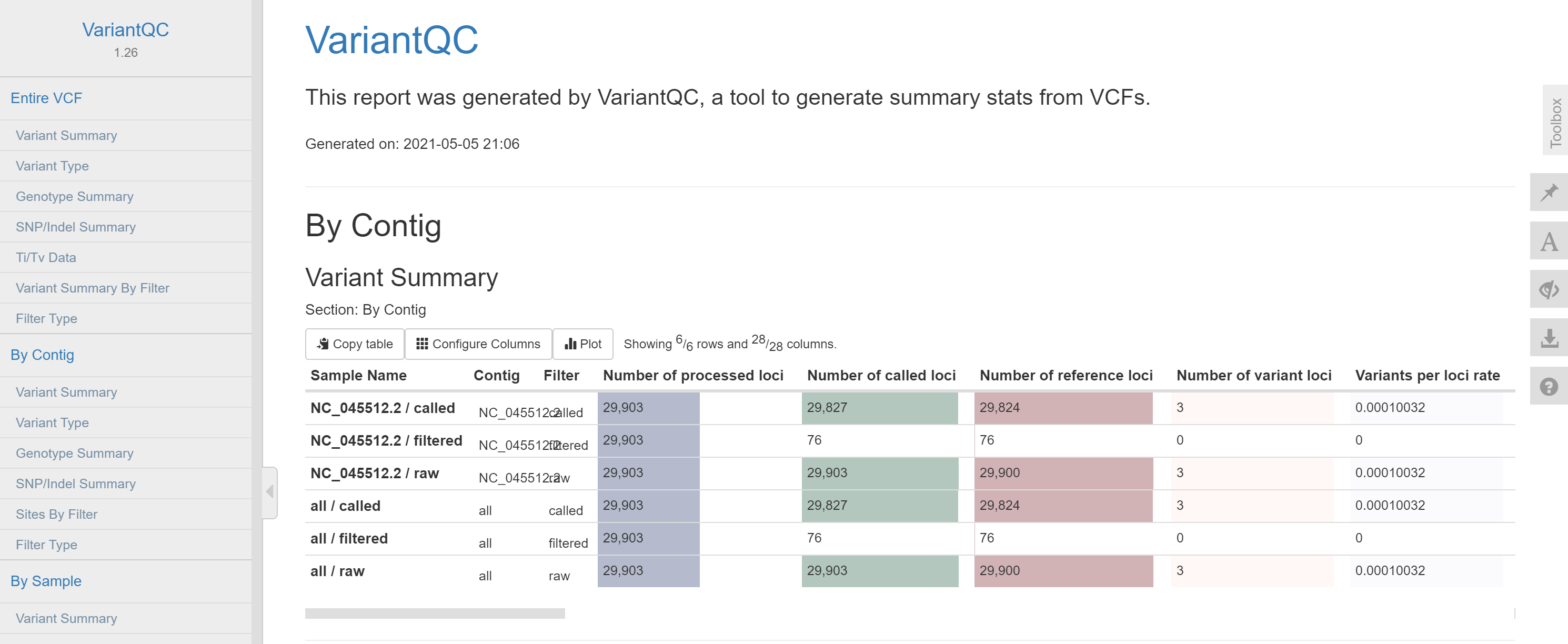
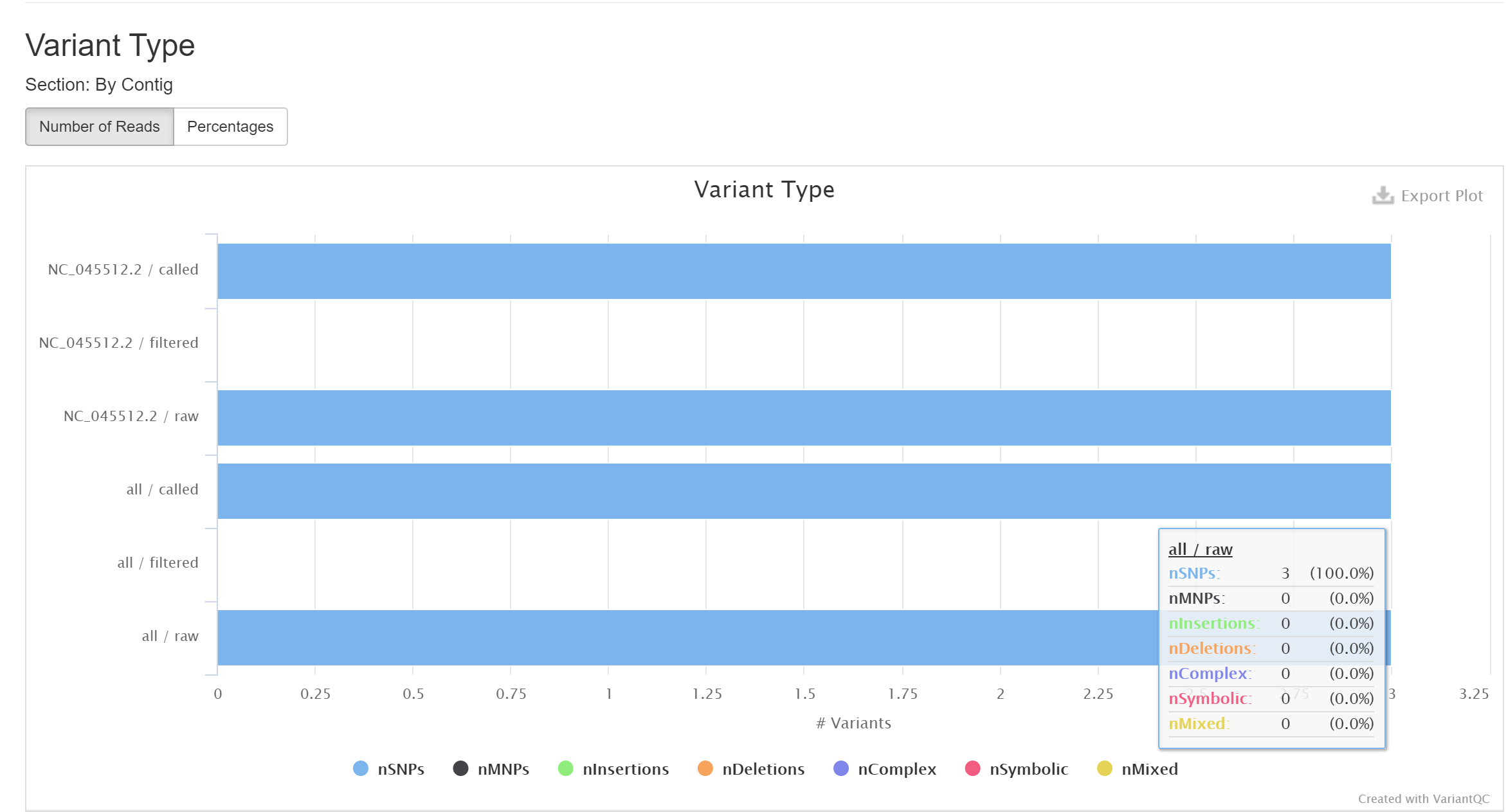
The report demonstrates that there are three mutation sites in the first genome. They are all point mutation.
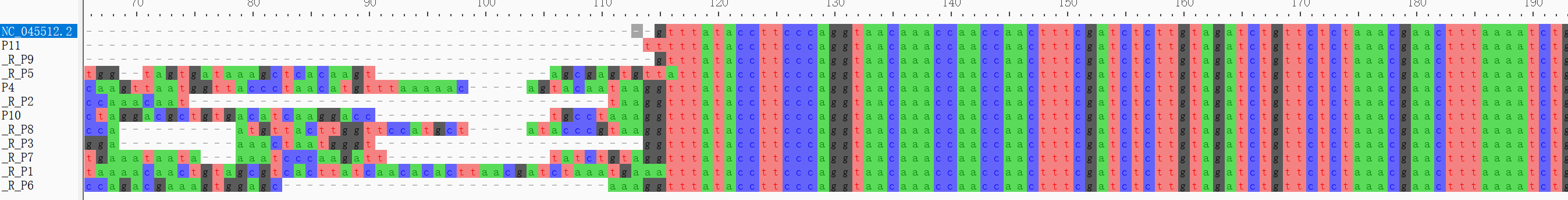
- Identify mutations in the viral genomes by comparing to the
**NC_045512.2**ref genome.

若有收获,就点个赞吧
0 人点赞
