MAFFT
Multiple alignment program for amino acid or nucleotide sequences.
Options:
$ mafft --help
High speed:% mafft in > out% mafft --retree 1 in > out (fast)High accuracy (for <~200 sequences x <~2,000 aa/nt):% mafft --maxiterate 1000 --localpair in > out (% linsi in > out is also ok)% mafft --maxiterate 1000 --genafpair in > out (% einsi in > out)% mafft --maxiterate 1000 --globalpair in > out (% ginsi in > out)If unsure which option to use:% mafft --auto in > out--clustalout # Output: clustal format, default: fasta--thread # Number of threads (if unsure, --thread -1)--inputorder # Output order: same as input. Default: on--reorder # Output order: aligned. Default: off (inputorder)--adjustdirection # Adjust the direction according to the first sequence
Online version: https://mafft.cbrc.jp/alignment/server/
LAST hits (score>39) between the top sequence and the others.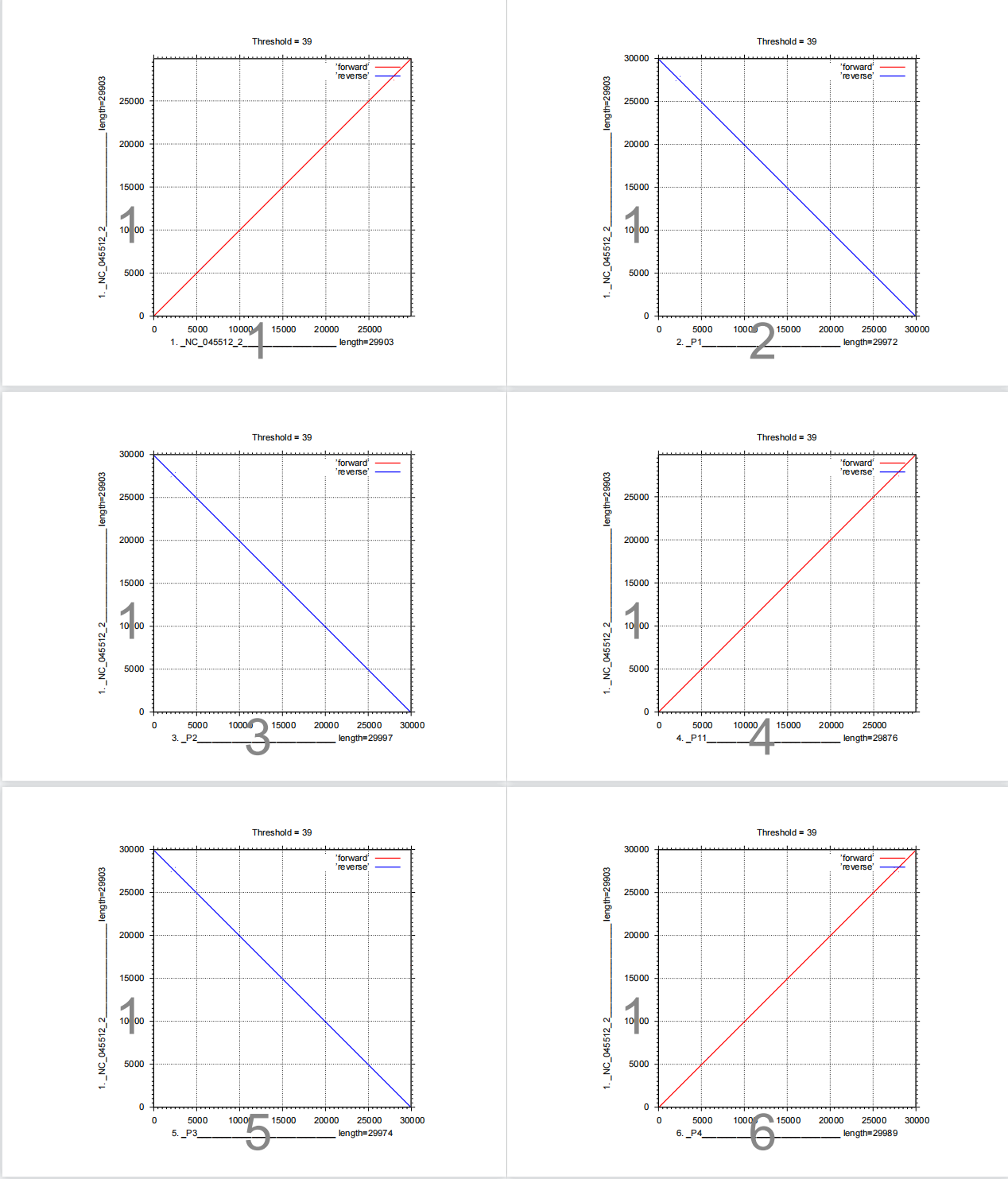
The assembled genome sequence2, 3 and 5 are reverse compared to NC_045512.2. Thus when aligning these sequences, --adjustdirection option should be added.
Manual: https://mafft.cbrc.jp/alignment/software/manual/manual.html
iqtree
IQ-TREE takes as input a multiple sequence alignment and will reconstruct an evolutionary tree that is best explained by the input data.
Options:
-s # Specify input alignment file in PHYLIP, FASTA, NEXUS, CLUSTAL or MSF format.-m # Specify the model name to use during the analysis-redo # Redo the entire analysis no matter if it was stopped or successful. WARNING: This option will overwrite all existing output files.--prefix # change the output files' prefix
Output files:
example.phy.iqtree: the main report file that is self-readable.example.phy.treefile: the ML tree in NEWICK format, which can be visualized by any supported tree viewer programs like FigTree or iTOL.example.phy.log: log file of the entire run (also printed on the screen).example.phy.ckp.gz: IQ-TREE automatically performs check pointing to resume an interrupted analysis. During the program run, IQ-TREE periodically wrote to disk a checkpoint fileexample.phy.ckp.gz. This checkpoint file is used to resume an interrupted run. If the run did not finish, invoking IQ-TREE again with the very same command line will recover the analysis from the last stopped point.- If the run successfully completed, running again will issue an error message:
If you really want to re-run the analysis and overwrite all previous output files, useERROR: Checkpoint (example.phy.ckp.gz) indicates that a previous run successfully finishedUse `-redo` option if you really want to redo the analysis and overwrite all output files.
-redooption:Manual:
http://www.iqtree.org/doc/Tutorial
https://github.com/iqtree/iqtreeiTOL
iTOL is an online tool for the display, annotation and management of phylogenetic and other trees.
- If the run successfully completed, running again will issue an error message:
iTOL: Interactive Tree Of Life (embl.de)
Pipeline
Alignment
mafft --reorder --adjustdirection --auto --clustalout --thread 4 ~/SARS_CoV_2/mafft/sequences112.fasta > output.aln
View
.alnfile usingAliView
The sequences adjusted direction were added a prefix ‘R‘.Infer maximum-likelihood tree using
**GTR+I+G**model.iqtree -s /public/home/ykk/SARS_CoV_2/mafft/output.aln -m GTR+I+G

Visualize
**.treefile**by tree viewer programs iTOL.

- Identify mutations in the viral genomes by comparing to the
**NC_045512.2**ref genome.mafft --reorder --adjustdirection --auto --clustalout --thread 4 ~/SARS_CoV_2/mafft/sequences12.fasta > output1.aln


若有收获,就点个赞吧
0 人点赞
