1. FASTQ files
FASTQfiles are compressed and created with the extension_*.fq.gz._
View the
FASTQfile$ zless -S P3-VERO-P3-1-vero_L4_1.fq.gz | head -n 4
@A00821:275:HWMMWDSXX:4:1101:1298:1016 1:N:0:ATTACTCG+TAGATCGCGCTTCTCATTAGAGATAATAGATGGTAGAATGTAAAAGGCACTTTTACACTTTTTAAGCACTGTCTTTGCCTCCTCTACAGTGTAACCATTTAAACCCTGACCCGGGTAAGTGGTTATATAATTGTCTGTTGGCACTTTTCTCAAAGCTT+FFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFF::FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
Each entry in a
FASTQfile consists of four lines:A sequence identifier with information about the sequencing run and the cluster. This line always starts with
@.ATTACTCG+TAGATCGC # index sequence
The sequence (the base calls; A, C, T, G and N).
- A separator, which is simply a plus (+) sign.
- The base call quality scores. These are
Phred+33 encoded, using ASCII characters to represent the numerical quality scores.- Relationship Between Sequencing Quality Score and Base Call Accuracy.

| Quality Score | Probability of Incorrect Base Call | Inferred Base Call Accuracy |
|---|---|---|
10 (Q10) |
1 in 10 | 90% |
20 (Q20) |
1 in 100 | 99% |
30 (Q30) |
1 in 1000 | 99.9% |
- **Higher Q scores **indicate a smaller probability of error.
2. Fastp
Fastpis a tool designed to provide fast all-in-one preprocessing forFastQfiles.
Fastpcan do comprehensive quality profiling for both before and after filtering data, meanwhile, it removes low-quality reads and bases. Most of thefastp‘s functions do not need to enter too many parameters. Some features are turned on by default, otherwise can be turned off with parameters.
2.1 Common Options:
-i <read1 input file name>-o <read1 output file name>-I <read2 input file name>-O <read2 output file name># Fastp supports the input and output of *.gz.-h <html format report name>-j <json format report name>-w <int> # --threads
Fastp supports both single-end (SE) and paired-end (PE) input/output.
# paired-endfastp -i ~/SARS_CoV_2/raw_data/P3-VERO-P3-1-vero_L4_1.fq.gz \-o ~/SARS_CoV_2/clean_data/P3-VERO-P3-1-vero_L4_1.fq.gz \-I ~/SARS_CoV_2/Fasta_file/P3-VERO-P3-1-vero_L4_2.fq.gz \-O ~/SARS_CoV_2/clean_data/P3-VERO-P3-1-vero_L4_2.fq.gz \-w 4 \--html 1.html \--json 1.json
2.2 Result
.fq.gzfileRaw data:
@A00821:275:HWMMWDSXX:4:1101:1298:1016 1:N:0:ATTACTCG+TAGATCGCGCTTCTCATTAGAGATAATAGATGGTAGAATGTAAAAGGCACTTTTACACTTTTTAAGCACTGTCTTTGCCTCCTCTACAGTGTAACCATTTAAACCCTGACCCGGGTAAGTGGTTATATAATTGTCTGTTGGCACTTTTCTCAAAGCTT+FFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFF::FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF@A00821:275:HWMMWDSXX:4:1101:1479:1016 1:N:0:ATTACTCG+TAGATCGCGCGTGTTTCTTCTGCATGTGCAAGCATTTCTCGCAAATTCCAAGAAACAGTTCCAAGAATTTCTTGCTTCTCATTAGAGATAATAGATGGTAGAATGTAAAAGGCACTTTTACACTTTTTAAGCACTGTCTTTGCCAGATCGGAAGAGCA+FFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFF:F:FFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFF:FFFFF
Clean data:
@A00821:275:HWMMWDSXX:4:1101:1298:1016 1:N:0:ATTACTCG+TAGATCGCGCTTCTCATTAGAGATAATAGATGGTAGAATGTAAAAGGCACTTTTACACTTTTTAAGCACTGTCTTTGCCTCCTCTACAGTGTAACCATTTAAACCCTGACCCGGGTAAGTGGTTATATAATTGTCTGTTGGCACTTTTCTCAAAGCTT+FFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFF::FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF@A00821:275:HWMMWDSXX:4:1101:1479:1016 1:N:0:ATTACTCG+TAGATCGCGCGTGTTTCTTCTGCATGTGCAAGCATTTCTCGCAAATTCCAAGAAACAGTTCCAAGAATTTCTTGCTTCTCATTAGAGATAATAGATGGTAGAATGTAAAAGGCACTTTTACACTTTTTAAGCACTGTCTTTGCC+FFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFF:F:FFFFFFFFFFFFFF:FFFFFFFFFFFFF
the second read was trimmed its adapter,
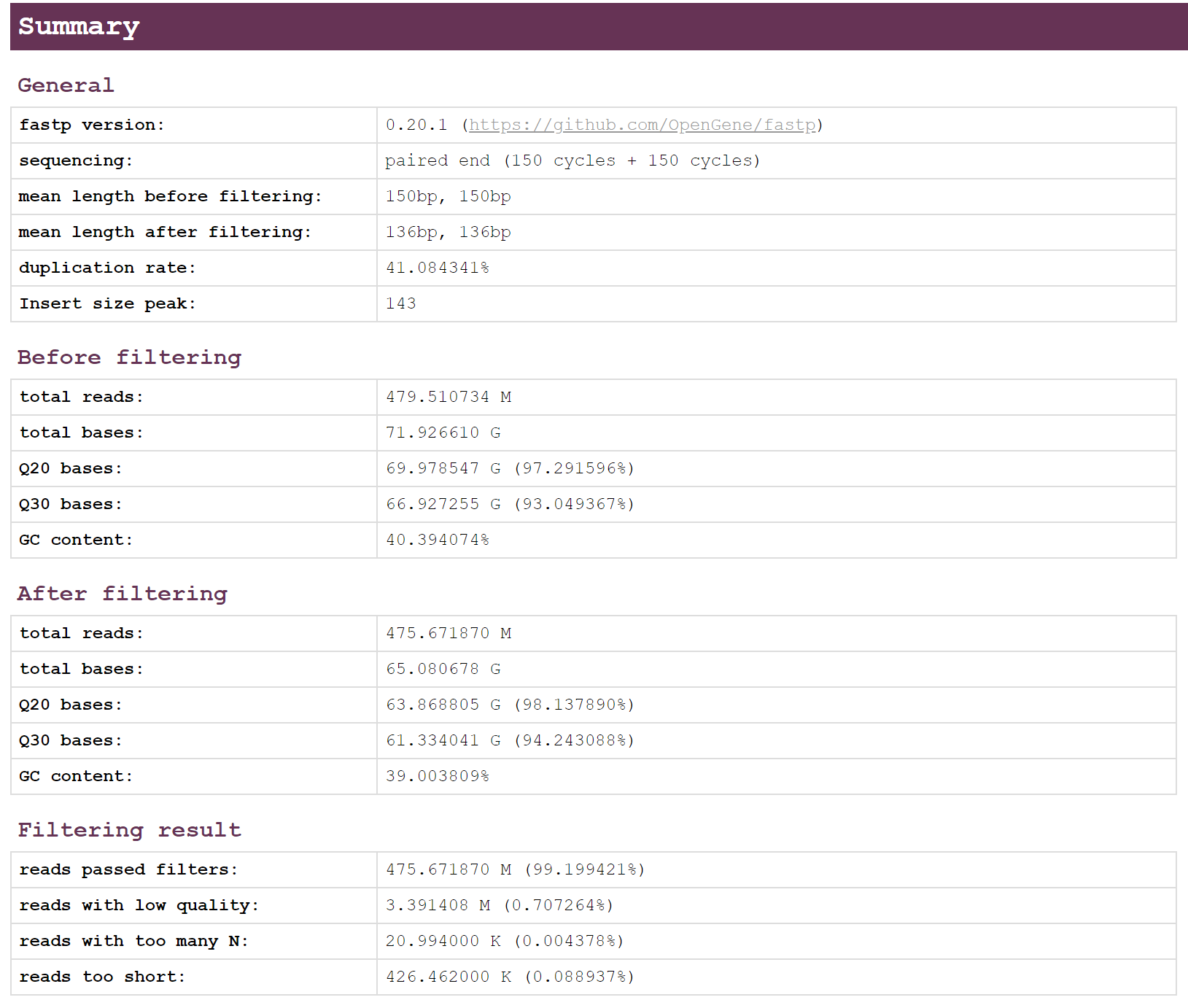
AGATCGGAAGAGCA.**.html**report- The summary shows statistics before and after filtering
- Adapters
Adapter sequences can be automatically detected for both PE/SE data.
3. More Information:
https://github.com/OpenGene/fastp
https://blog.csdn.net/twocanis/article/details/109681242

若有收获,就点个赞吧
0 人点赞
