MetaQUAST
MetaQUAST evaluates and compares metagenome assemblies based on alignments to close references. MetaQUAST can be fed with multiple assemblies, thus is perfect for comparison. MetaQUAST is distributed within QUAST package since version 2.2.
Options:
-o <dirname> # Directory to store all result files [default: quast_results/results_<datetime>]-r <filename,filename,...> # Comma-separated list of reference genomes or directory with reference genomes-m <int> # Lower threshold for contig length [default: 500]-t <int> # Maximum number of threads [default: 25% of CPUs]
manual:
$ conda activate rna$ metaquast.py --help
- Usage:
python /public/home/ykk/miniconda3/envs/covid/bin/metaquast.py [options] <files_with_contigs>
Evaluate and compare metagenome assemblies
scaffolds.fasta/contigs.fasta
**scaffolds.fasta**and**contigs.fasta**are no difference in the assembly results of this project.
python ~/miniconda3/envs/covid/bin/metaquast.py scaffolds.fasta contigs.fasta -r ~/SARS_CoV_2/mapping/ref/Wuhan-Hu-1.fasta -t 4 -o result_two
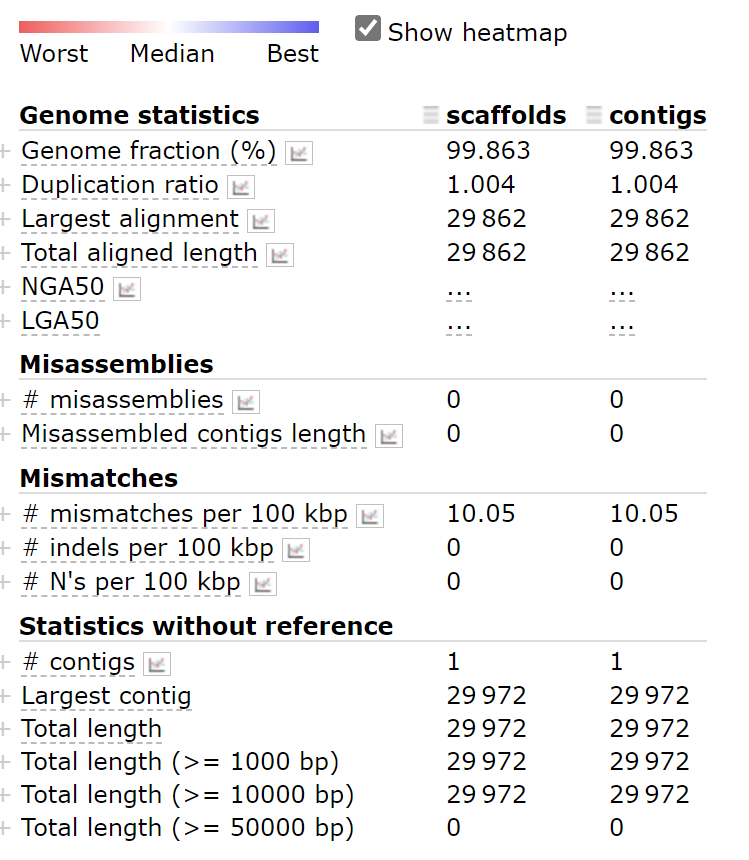
options: —meta/—isolate/—sc
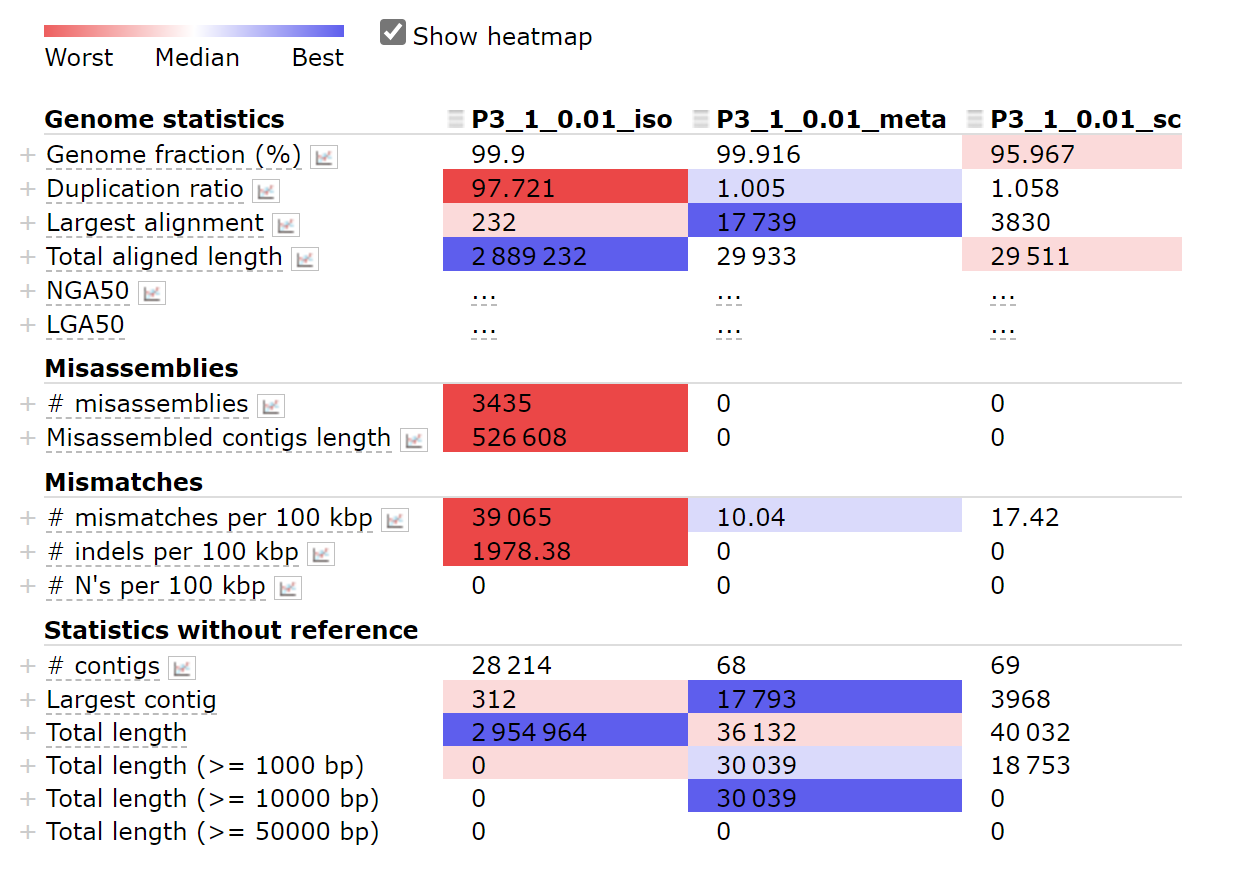
Using option —meta works best.
python ~/miniconda3/envs/covid/bin/metaquast.py P3-1_0.01_iso.fasta P3-1_0.01_meta.fasta P3-1_0.01_sc.fasta -m 0 -r ~/SARS_CoV_2/mapping/ref/Wuhan-Hu-1.fasta -t 4 -o result_com0.01
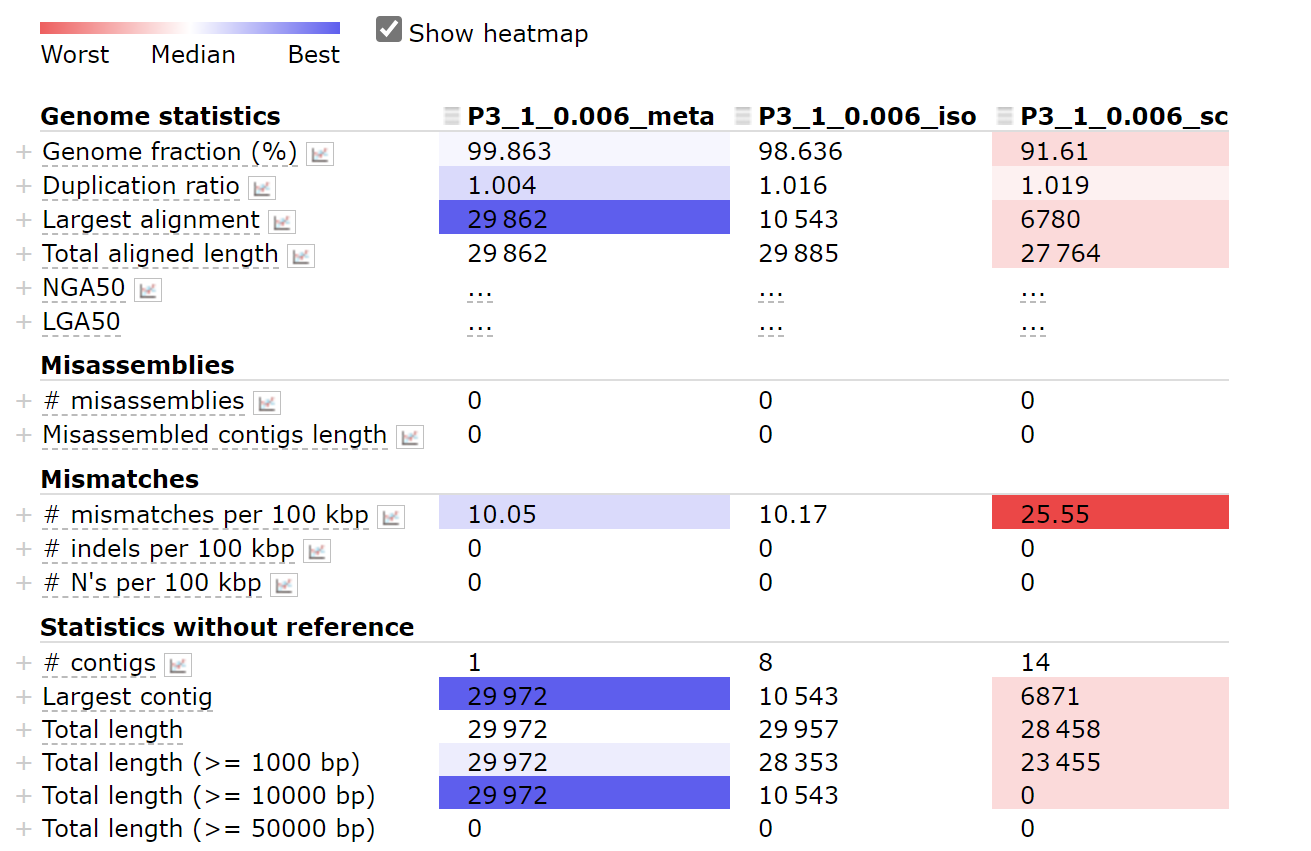
python ~/miniconda3/envs/covid/bin/metaquast.py P3-1_0.006_meta.fasta P3-1_0.006_iso.fasta P3-1_0.006_sc.fasta -r ~/SARS_CoV_2/mapping/ref/Wuhan-Hu-1.fasta -t 4 -o result_com0.006
-s
For the 9th sequence, using parameter 0.006 works best.
python ~/miniconda3/envs/covid/bin/metaquast.py P3-1_0.006_meta.fasta P3-1_0.01_meta.fasta P3-1_0.1_meta.fasta -r ~/SARS_CoV_2/mapping/ref/Wuhan-Hu-1.fasta -t 4 -o result_part
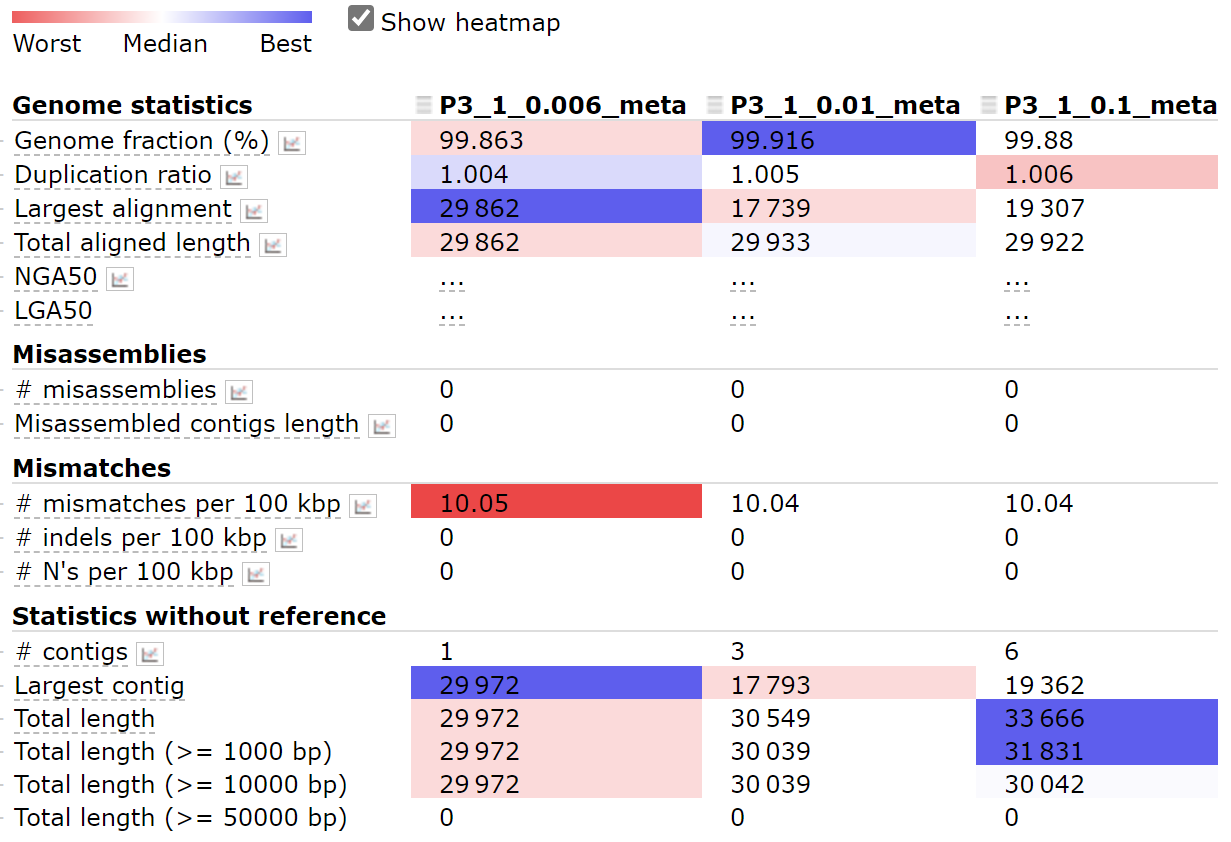
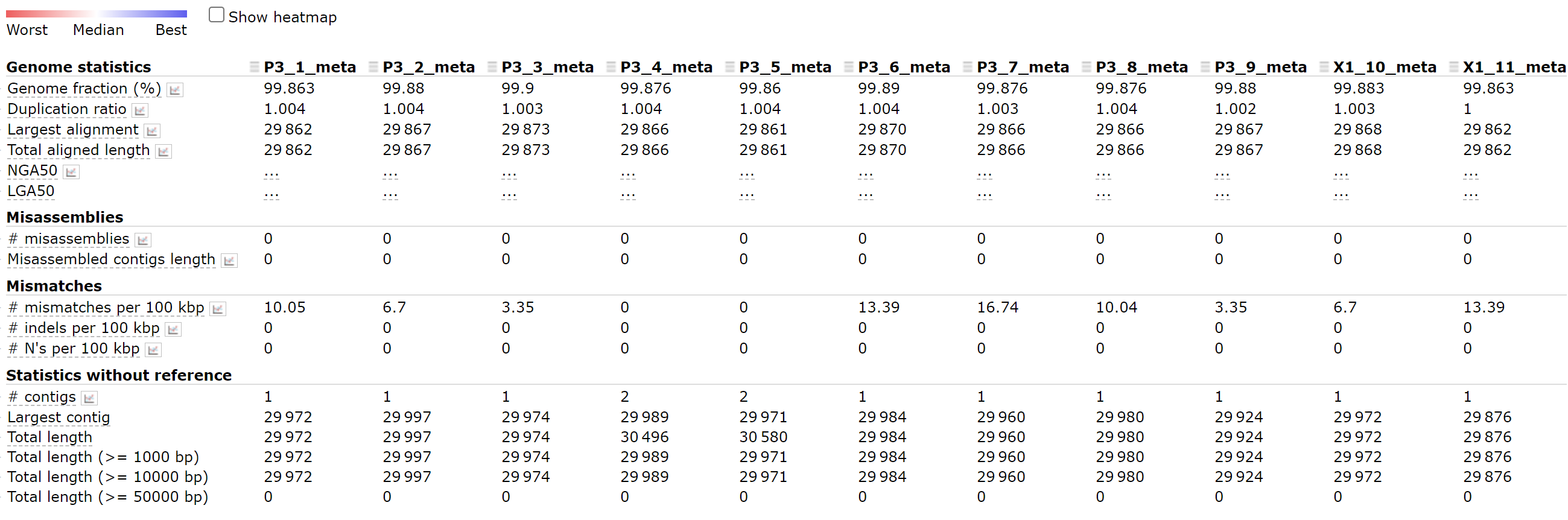
1-11 assemblies
python ~/miniconda3/envs/covid/bin/metaquast.py P3-1_meta.fasta P3-2_meta.fasta P3-3_meta.fasta P3-4_meta.fasta P3-5_meta.fasta P3-6_meta.fasta P3-7_meta.fasta P3-8_meta.fasta P3-9_meta.fasta X1-10_meta.fasta X1-11_meta.fasta -r ~/SARS_CoV_2/mapping/ref/Wuhan-Hu-1.fasta -t 16 -o result_last
More information:

若有收获,就点个赞吧
0 人点赞
