Since December 2019, the COVID-19 caused by a RNA coronavirus, SARS-CoV-2, has spread rapidly worldwide. Viruses can mutate and evolve rapidly during replication and spread. Therefore, studying the evolution and identifying the substitutions of SARS-COV-2 are essential for COVID-19 control. Previously, SARS-CoV-2 isolates from 11 patients were collected and the sequencing data were obtained by next-generation sequencing. In this workflow, the viral sequences were assembled and then compared with sequences in database to check its mutation. According to the phylogenetic analysis of SARS-CoV-2, the evolutionary history can be reconstructed to identify the virus infection source and transmission chain.
The Sequence File
These sequencing files were obtained from viral genome sequencing. These viruses were isolated from 11 COVID-19 patients. The sequence files we got are in FASTQ format. Then use the following code to view one of these files.
zless -S P3-VERO-P3-1-vero_L4_1.fq.gz | head -n 8
A
FASTQfile normally uses four lines for storing sequencing information per sequence.@A00821:275:HWMMWDSXX:4:1101:1298:1016 1:N:0:ATTACTCG+TAGATCGCGCTTCTCATTAGAGATAATAGATGGTAGAATGTAAAAGGCACTTTTACACTTTTTAAGCACTGTCTTTGCCTCCTCTACAGTGTAACCATTTAAACCCTGACCCGGGTAAGTGGTTATATAATTGTCTGTTGGCACTTTTCTCAAAGCTT+FFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFF::FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
- This first line always starts with a
@character and is followed by a sequence identifier. - The second line is the raw sequence letters.
- The last line encodes the quality values for the sequence in the second line. If p is the error probability, then:
 . Q scores are often represented as ASCII characters.
. Q scores are often represented as ASCII characters.
Workflow
1. Install software
During the analysis process, we need many tools to analyze our data. These tools can be downloaded usingConda.Condais a powerful package manager and environment manager.Condaquickly installs, runs and updates packages and their dependencies, as well, it easily creates, saves, loads, and switches between environments. ```shell———————-Create a environment
conda create -y -n covid python=3Activate covid environment
conda activate covid
———————-Install software in ‘covid’ environment
conda install -y fastp trim-galore bwa samtools sambamba spades quast pilon mafft iqtree
<a name="Os74W"></a>### 2. Create and manage directories```shellmkdir SARS_CoV_2cd SARS_CoV_2mkdir {raw_data,qc,clean_data,mapping,sort,subsample,fastq,genome,quast,Pilon,mafft}
3. Quality Control
Before further analysis, low-quality regions and adaptors should be trimmed and removed by Fastp or trim_galore from raw sequencing data. This step is important to avoid issues in downstream analyses such as variant analysis or assembly.
cd ~/SARS_CoV_2/qc##---------------fastpfor file in $(ls ~/SARS_CoV_2/raw_data/*_1.fq.gz)dofullname=${file##*/}fastp -i ${file%1.*}1.fq.gz \-o ~/SARS_CoV_2/clean_data/${fullname%1.*}1.fq.gz \-I ${file%1.*}2.fq.gz \-O ~/SARS_CoV_2/clean_data/${fullname%1.*}1.fq.gz \-w 4 \--html ${fullname%_1.*}.html \--json ${fullname%_1.*}.jsondoneor##---------------trim_galorefor file in $(ls ~/SARS_CoV_2/raw_data/*_1.fq.gz)dotrim_galore --output_dir ~/SARS_CoV_2/clean_data \--phred33 --paired \--length 75 --quality 30 --stringency 5 \${file%1.*}1.fq.gz ${file%1.*}2.fq.gzdone
In this period, we chose Fastp to filter the raw data. Fastp per-cycle quality profiles, per-cycle base contents, adapter trimming results, and k-mer counts. These values will be merged after all reads are processed, and a reporter will generate reports in HTML and JSON formats. Fastp reports statistical values for pre-filtering and post-filtering data to facilitate comparisons of changes in data quality after filtering is complete.
- HTML report
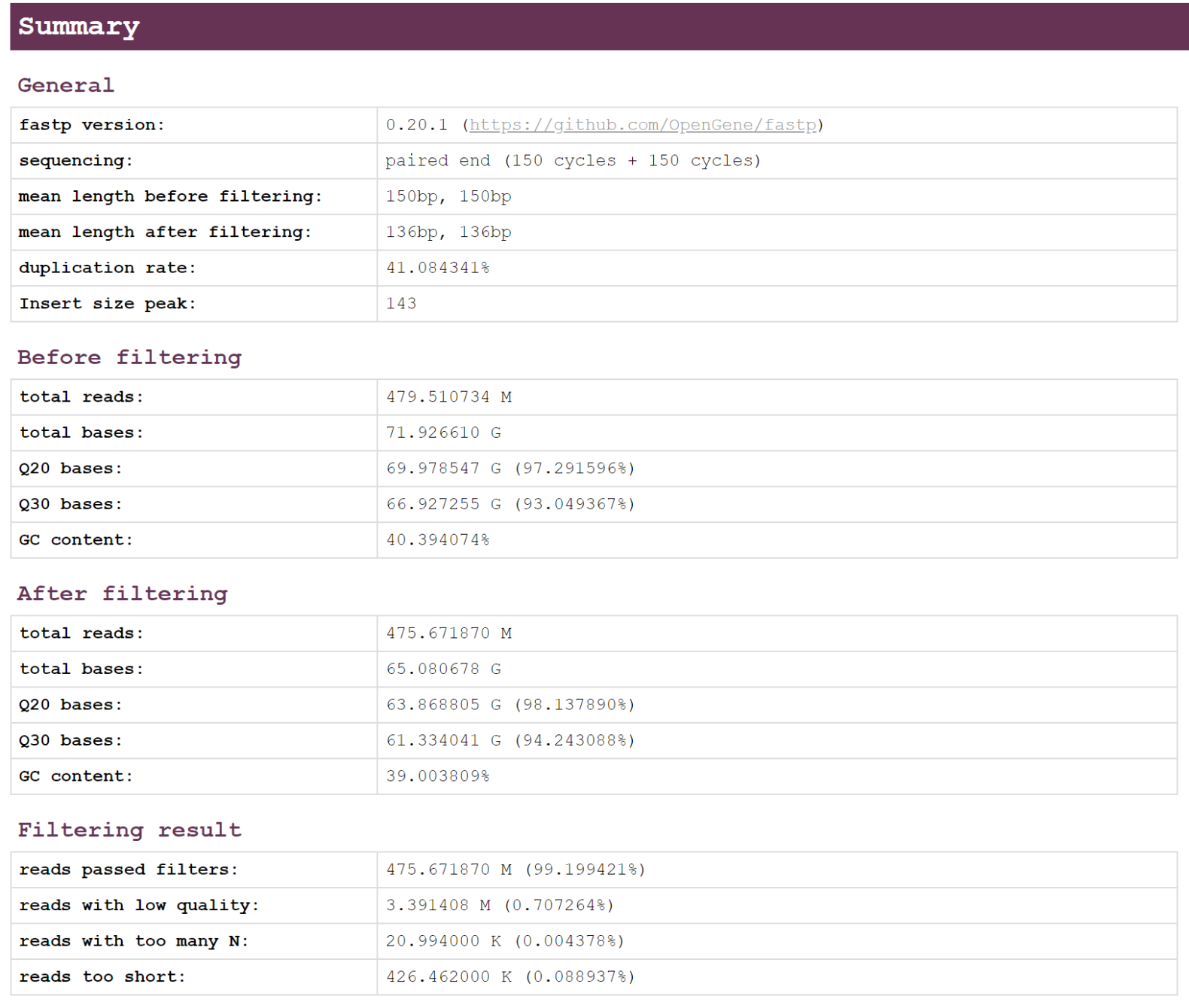
4. Map sequence
Paired reads were aligned to the 29903nt Wuhan reference genome (NCBI NC_045512.2) using BWA.
##---------------Build indexcd ~/SARS_CoV_2/mappingmkdir ref# Download the reference genome from the URL: https://www.ncbi.nlm.nih.gov/nuccore/NC_045512.2?report=fasta, and rename the fasta file to 'Wuhan-Hu-1.fasta'bwa index Wuhan-Hu-1.fasta
##---------------Map sequence by bwa-memcd ~/SARS_CoV_2/mappingfor file in $(ls ~/SARS_CoV_2/clean_data/*_1.fq.gz)dofullname=${file##*/}INDEX=~/SARS_CoV_2/mapping/ref/Wuhan-Hu-1.fastabwa mem -t 4 -M $INDEX \${file%_1.*}_1.fq.gz \${file%_1.*}_2.fq.gz \> ~/SARS_CoV_2/mapping/${fullname%_1.*}.sam \2> ~/SARS_CoV_2/mapping/${fullname%_1.*}.logdonefor file in $(ls ~/SARS_CoV_2/mapping/*.sam)dofullname=${file##*/}samtools view -b -F 12 $file > ${file%.sam*}.bam &&\samtools sort -@ 4 -l 9 ${file%.sam*}.bam -o ~/SARS_CoV_2/sort/${fullname%.sam*}_sort.bam &&\samtools index ~/SARS_CoV_2/sort/${fullname%.sam*}_sort.bam ~/SARS_CoV_2/sort/${fullname%.sam*}_sort.bam.baidone##---------------Count the number of alignmentscd ~/SARS_CoV_2/sortfor file in $(ls ~/SARS_CoV_2/sort/*.bam)dofullname=${file##*/}samtools flagstat $file > ${fullname%.bam*}_flagstat.txtdone##---------------Calculate the sequencing depthfor file in $(ls ~/SARS_CoV_2/sort/*.bam)dofullname=${file##*/}samtools depth -d 0 $file > ${fullname%.bam*}_depth.txtdone
5. Assemble genome
SPAdes is an assembly toolkit mainly used for small genomes, so it’s a good choice for assembling viral genomes. SPAdes contains various assembly pipelines. In this step, we chose the pipeline, metaSPAdes. In the next part, we would compare the different effects between the metaSPAdes and others.
##---------------Subsample readscd ~/SARS_CoV_2/subsamplefor file in $(ls ~/SARS_CoV_2/sort/*.bam)dofullname=${file##*/}sambamba view -h -s 0.001 $file -o ~/SARS_CoV_2/subsample/${fullname%-vero*}-0.001.bamdone##---------------Sort and build indexfor file in $(ls ~/SARS_CoV_2/subsample/*.bam)dosamtools sort -@ 4 -l 9 $file -o ${file%.bam}_sort.bam &&\samtools index ${file%.bam}_sort.bam ${file%.bam}_sort.bam.baidone##---------------Convert the BAM to FASTQcd ~/SARS_CoV_2/fastqfor file in $(ls ~/SARS_CoV_2/subsample/*_sort.bam)dofullname=${file##*/}samtools fastq $file > ~/SARS_CoV_2/fastq/${fullname%_sort.bam*}.fqdone##---------------Assemblecd ~/SARS_CoV_2/genomefor file in $(ls ~/SARS_CoV_2/fastq/*.fq)dofullname=${file##*/}spades.py --meta --12 $file -o ~/SARS_CoV_2/genome/${fullname%.fq*}_meta.fastadone
6. Evaluate assembly
To evaluate the effect of assembly, we used MetaQUAST to compare the assembled genome with the reference genome(NCBI NC_045512.2).
- N50
For genome assembly results, we usually evaluate them by N50 and other indicators. For an assembled sequence, the sequences are sorted from the largest to the smallest according to their length, and then the lengths are accumulated starting from the first sequence until the cumulative length exceeds 50% of the total length. At this point, the length of the last contig accumulated is the length of N50. The schematic diagram is as follows: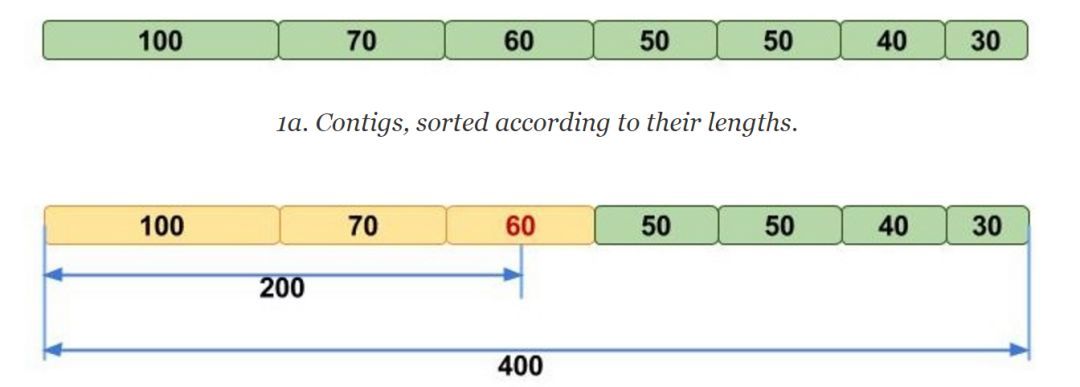
a. scaffolds.fasta/contigs.fasta
 cbin
cbin
python ~/miniconda3/envs/covid/bin/metaquast.py scaffolds.fasta contigs.fasta -r ~/SARS_CoV_2/mapping/ref/Wuhan-Hu-1.fasta -t 4 -o result_version
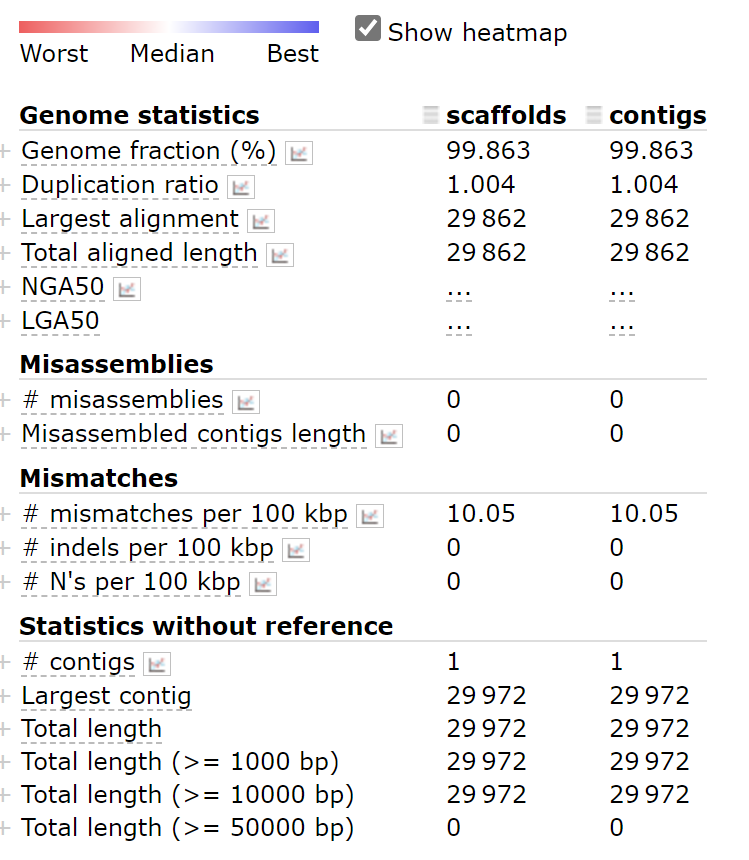
The N50 is not shown in the screenshot, but according to the definition of N50, assembled genomes can be measured by the ratio of the largest alignment to the total aligned length. The higher the ratio, the better. scaffolds.fasta and contigs.fasta have no difference in the assembly results. We can pick any one of them for subsequent analysis.
b. Different pipelines in MAFFT
When we assemble a genome using MAFFT, there are three options(—meta/—sc/—iso) we can use. 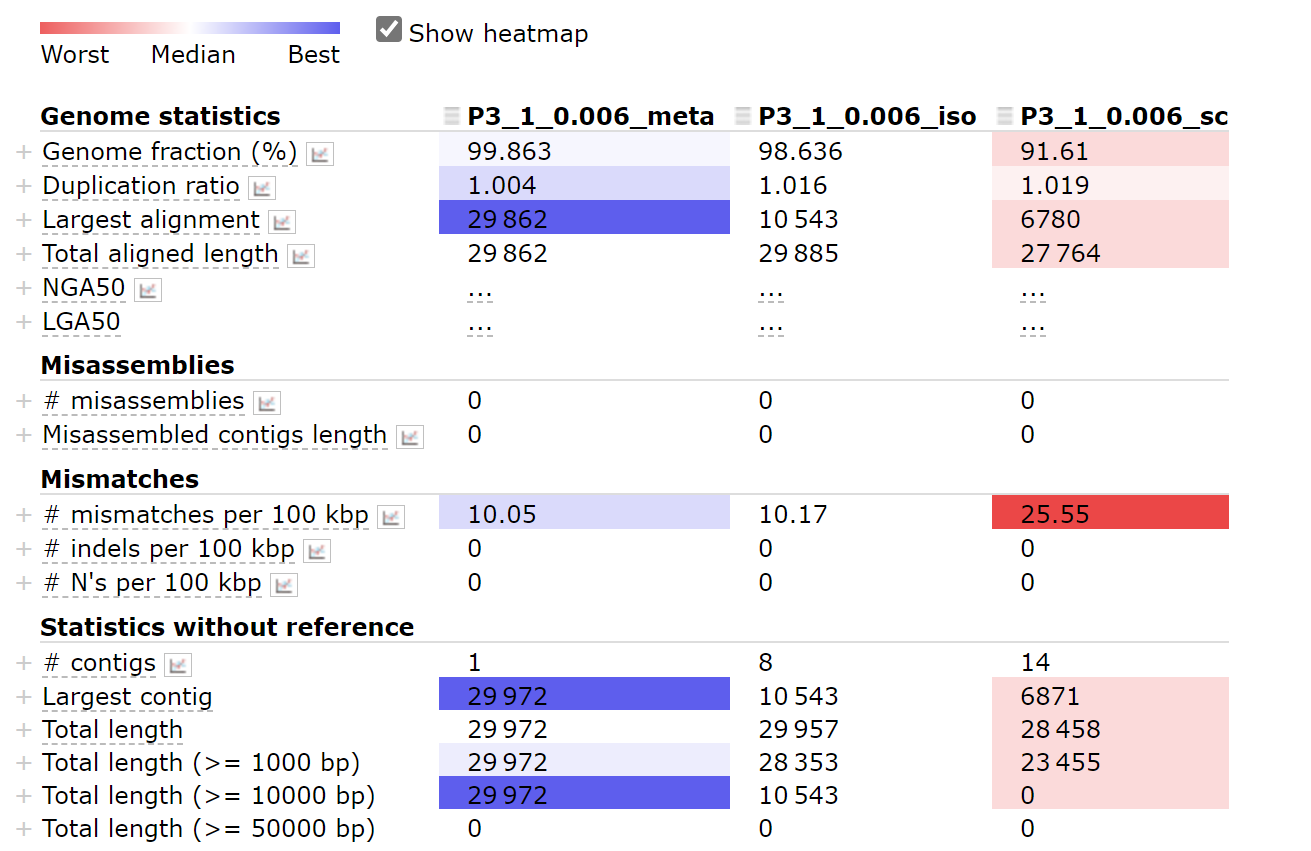
When using the metaSPAdes pipeline, the ratio of the largest alignment to the total aligned length is higher.
c. Different subsampling coverage
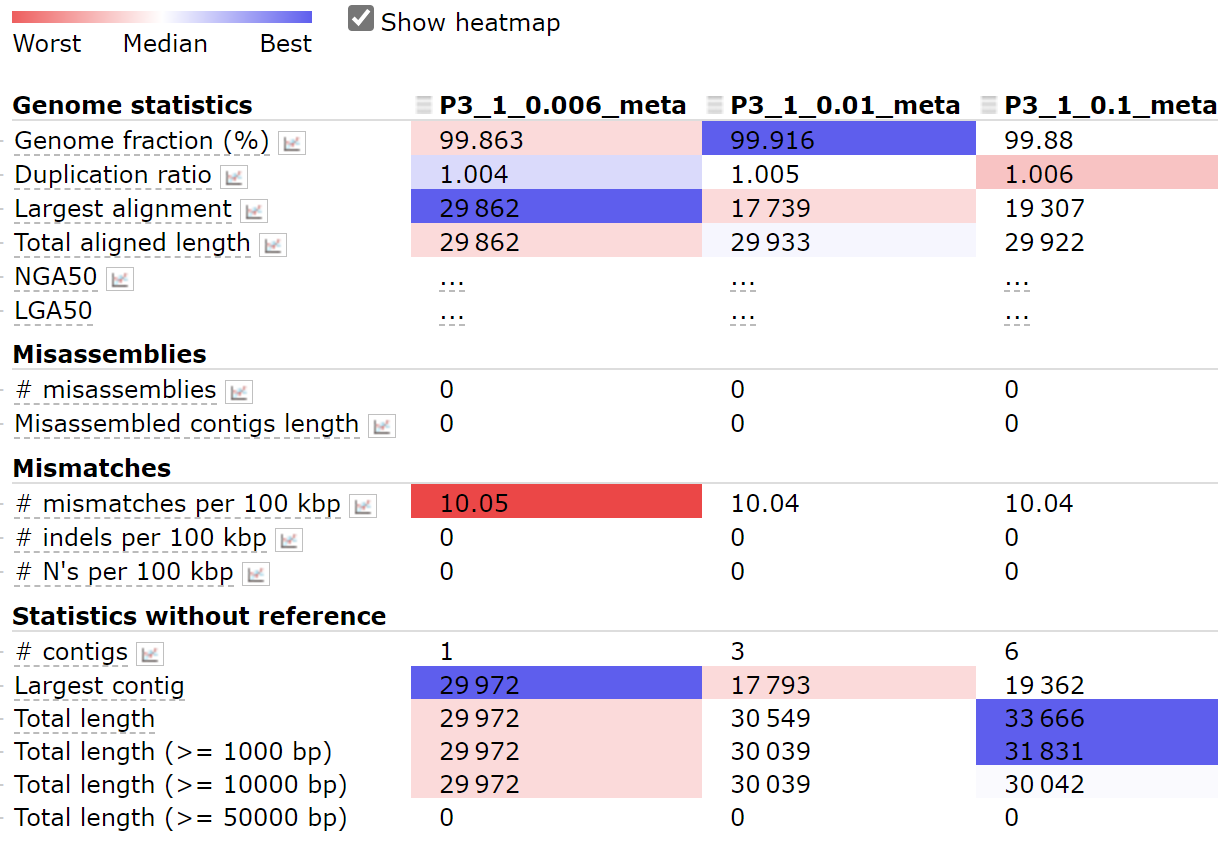
Before assembling the genome, we subsampled our reads to get better results. Using different subsampling coverage makes a difference. For the first genome, 0.006 is a better fraction. However, for other genomes, it could be another one.
7. Improve assemblies
Pilon is a software tool that can be used to automatically improve draft assemblies by comparing the raw sequenced data with the assembled genome.
##---------------Build indexcd ~/SARS_CoV_2/Pilonfor i in {1..11}domkdir P3-${i}bwa index -p P3-${i}/P3-${i}_meta ~/SARS_CoV_2/genome/*${i}*.fastadone##---------------Align and sortfor i in {1..11}dobwa mem -t 20 ~/SARS_CoV_2/Pilon/P3-${i}/P3-${i}_meta \~/SARS_CoV_2/clean_data/*-${i}-*_1.fq.gz \~/SARS_CoV_2/clean_data/*-${i}-*_2.fq.gz \| samtools sort -@ 10 -O bam -o ./P3-${i}/P3-${i}_meta.bam &&\samtools index -@ 10 ./P3-${i}/P3-${i}_meta.bamdone##---------------Mark duplicationfor i in {1..11}dosambamba markdup -t 20 --hash-table-size=1000000 --overflow-list-size=1000000 ./P3-${i}/P3-${i}_meta.bam ./P3-${i}/P3-${i}_markdup.bamdone##---------------Filterfor i in {1..11}dosamtools view -@ 10 -q 30 -b -h ./P3-${i}/P3-${i}_markdup.bam > ./P3-${i}/P3-${i}_filter.bam &&\samtools index -@ 10 ./P3-${i}/P3-${i}_filter.bamdone##---------------Improve assembliesfor i in {1..11}dojava -Xmx16G -jar ~/miniconda3/envs/covid/share/pilon-1.23-3/pilon-1.23.jar \--genome ./P3-${i}/P3-${i}_meta.fasta \--frags ./P3-${i}/P3-${i}_filter.bam \--changes --fix all,breaks\--output ./P3-${i}/pilon_polished_${i} \--vcf &> ./P3-${i}/pilon${i}.logdone
8. Identify mutations
a. Find Variation Using Pilon
Input to Pilon consists of the NC_045512.2 genome in FASTA format along with the BAM files of sequencing reads aligned to the NC_045512.2 genome.
##---------------Identify mutationsfor i in {1..11}dojava -Xmx64G -jar ~/miniconda3/envs/covid/share/pilon-1.23-3/pilon-1.23.jar \--genome ~/SARS_CoV_2/mapping/ref/Wuhan-Hu-1.fasta \--frags ~/SARS_CoV_2/subsample/*${i}*_sort.bam \--changes \--output ${i}.fasta \--fix all,breaks\done
· View **.changes** File
The .changes file contains a space-delimited record of every change made in the assembly as instructed by the --fix option.
for i in {1..11}doecho ">${i}.fasta.changes" >> sum.txtcat ${i}.fasta.changes >>sum.txtdone
>1.fasta.changesNC_045512.2:11083 NC_045512.2_pilon:11083 G TNC_045512.2:23607 NC_045512.2_pilon:23607 G ANC_045512.2:26144 NC_045512.2_pilon:26144 G T>2.fasta.changesNC_045512.2:8782 NC_045512.2_pilon:8782 C TNC_045512.2:28144 NC_045512.2_pilon:28144 T C>3.fasta.changesNC_045512.2:16846 NC_045512.2_pilon:16846 G T>4.fasta.changes>5.fasta.changes>6.fasta.changesNC_045512.2:3885 NC_045512.2_pilon:3885 C ANC_045512.2:12778 NC_045512.2_pilon:12778 C TNC_045512.2:13270 NC_045512.2_pilon:13270 T CNC_045512.2:29449 NC_045512.2_pilon:29449 G T>7.fasta.changesNC_045512.2:1405 NC_045512.2_pilon:1405 T CNC_045512.2:9802 NC_045512.2_pilon:9802 G TNC_045512.2:19945 NC_045512.2_pilon:19945 A GNC_045512.2:25267 NC_045512.2_pilon:25267 C TNC_045512.2:27615 NC_045512.2_pilon:27615 C G>8.fasta.changesNC_045512.2:8782 NC_045512.2_pilon:8782 C TNC_045512.2:18060 NC_045512.2_pilon:18060 C TNC_045512.2:28144 NC_045512.2_pilon:28144 T C>9.fasta.changesNC_045512.2:22303 NC_045512.2_pilon:22303 T G>10.fasta.changesNC_045512.2:16114 NC_045512.2_pilon:16114 C TNC_045512.2:22205 NC_045512.2_pilon:22205 G C>11.fasta.changesNC_045512.2:22303 NC_045512.2_pilon:22303 T GNC_045512.2:27775 NC_045512.2_pilon:27775 T CNC_045512.2:27776 NC_045512.2_pilon:27776 T GNC_045512.2:27777 NC_045512.2_pilon:27777 G A
Through the changes file, we can find that there are 3 mutations in the first genome. More detailed information is stored in the 1.fasta.vcf file.
b. Find Variation Using MAFFT
cat ~/SARS_CoV_2/Pilon/P3-*/pilon_polished*.fasta ~/SARS_CoV_2/mapping/ref/Wuhan-Hu-1.fasta > sequences12.fastamafft --reorder --adjustdirection --auto --clustalout --thread 4 ~/SARS_CoV_2/mafft/sequences12.fasta > mut.aln
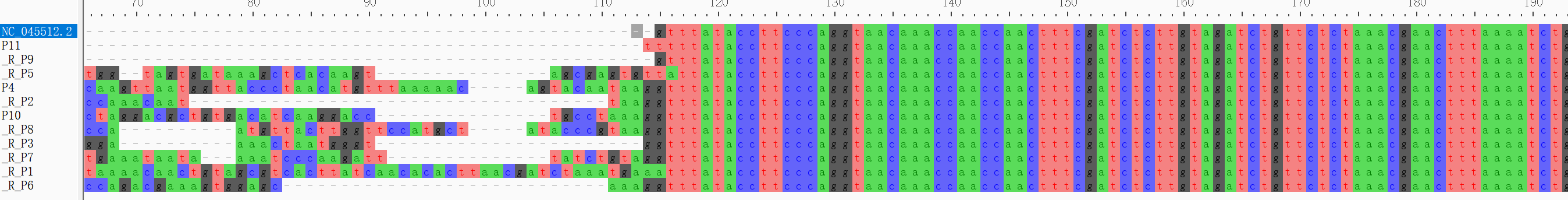
9. ML phylogenetic tree
a. Multiple sequence alignment
The assembled virus genomes was aligned with the previous genome using MAFFT. The previous genome sequences were obtained from the NCBI SARS-CoV-2 Resources(https://www.ncbi.nlm.nih.gov/sars-cov-2/) in fasta format.
cd ~/SARS_CoV_2/mafft# Download 100 SARS-CoV-2 genome sequences randomly from SARS-CoV-2 Database.cat sequences.fasta ~/SARS_CoV_2/Pilon/P3-*/pilon_polished*.fasta ~/SARS_CoV_2/mapping/ref/Wuhan-Hu-1.fasta > sequences112.fasta##---------------Multiple alignmentmafft --reorder --adjustdirection --auto --clustalout --thread 4 ./sequences112.fasta > output.aln

b. Construct an evolutionary tree
After alignment using MAFFT, the output.aln file can be used to construct an evolutionary tree using the GTR+I+G model in iqtree. Using their evolutionary tree, we can determine the chain of transmission in these 11 patients.
iqtree -s./output.aln -m GTR+I+G
c. Visualize .treefile by tree viewer programs iTOL.
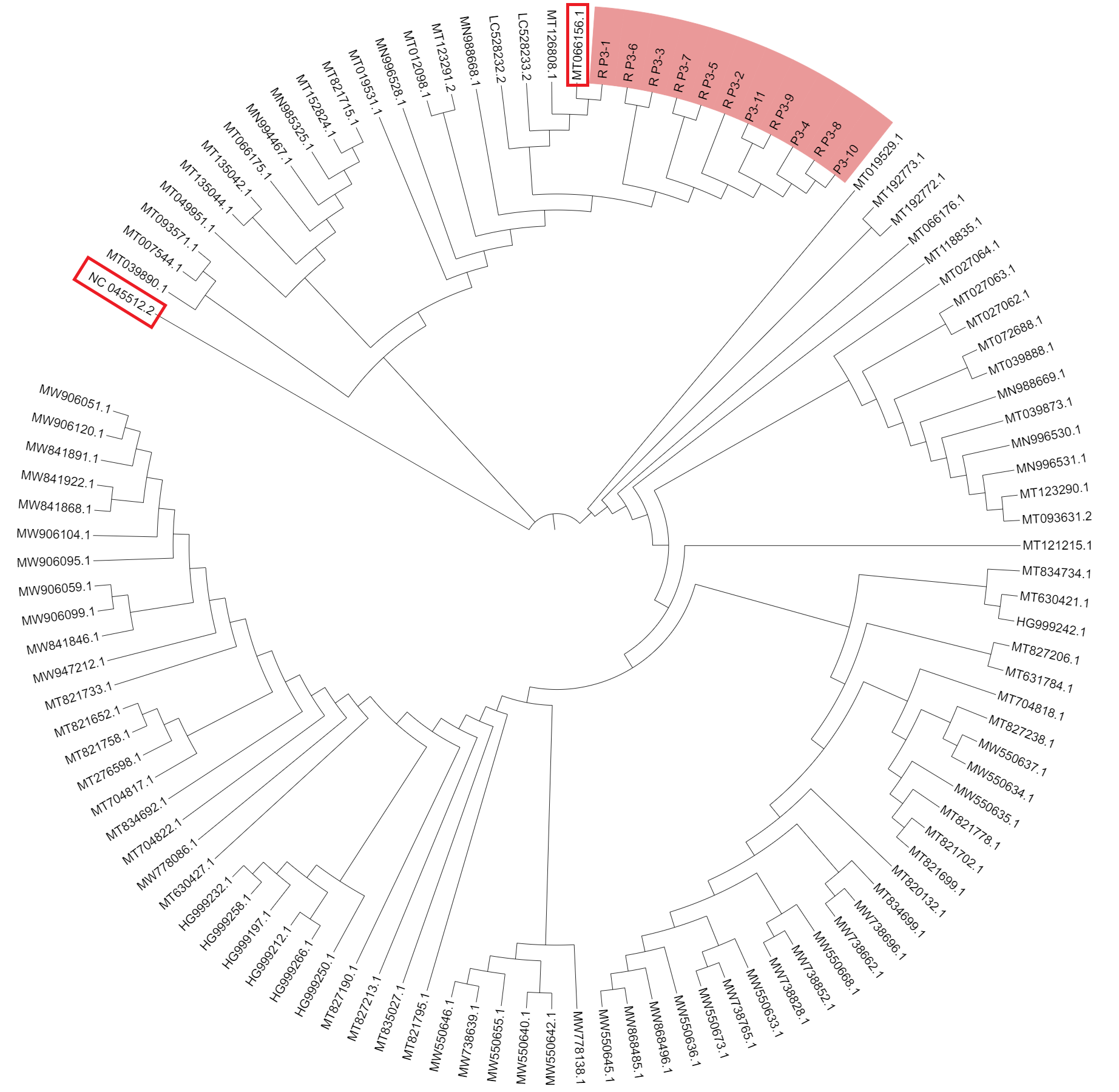
According to the evolutionary tree, the virus with genome ID MT066156.1 is most closely related to the 11 strains. MT066156.1 was isolated from the first case of COVID-19 in Italy. The patient came to Italy as tourist from Hubei province. They’re further away from the virus with genome ID NC045512.2. The 11 cases were indirectly infected by those people who linked to Huanan Seafood Market.

若有收获,就点个赞吧
0 人点赞
