简介
在发育过程中,细胞对刺激作出反应,并在整个生命过程中,从一种功能“状态”过渡到另一种功能“状态”。不同状态的细胞表达不同的基因,产生蛋白质和代谢物的动态重复序列,从而完成它们的工作。当细胞在状态之间移动时,它们经历一个转录重组的过程,一些基因被沉默,另一些基因被激活。这些瞬时状态通常很难描述,因为在更稳定的端点状态之间纯化细胞可能是困难的或不可能的。单细胞RNA-Seq可以使您在不需要纯化的情况下看到这些状态。然而,要做到这一点,我们必须确定每个cell在可能的状态范围内的位置。
Monocle介绍了利用RNA-Seq进行单细胞轨迹分析的策略。Monocle不是通过实验将细胞纯化成离散状态,而是使用一种算法来学习每个细胞必须经历的基因表达变化序列,作为动态生物学过程的一部分。一旦它了解了基因表达变化的整体“轨迹”,Monocle就可以将每个细胞置于轨迹中的适当位置。然后,您可以使用Monocle的微分分析工具包来查找在轨迹过程中受到调控的基因,如查找作为伪时间函数变化的基因一节所述。如果这个过程有多个结果,Monocle将重建一个“分支”轨迹。这些分支与细胞的“决策”相对应,Monocle提供了强大的工具来识别受它们影响的基因,并参与这些基因的形成。在分析单细胞轨迹中的分支的小节中,您可以看到如何分析分支。
monocle 能做的不只是拟时分析,或者说为了做拟时分析他也做了sc-rna-seq的基本分析流程:数据读入,均一化,降维(PCA,umap,tsne,),聚类,marker基因筛选以及可视化函数。在新的学习中我们发现monocle能做的远不只这些,例如用shiny开发了web程序,更加用户友好;借助garnett包可以做细胞定义——-monocle已经是一个sc-rna-seq数据分析的工具箱。
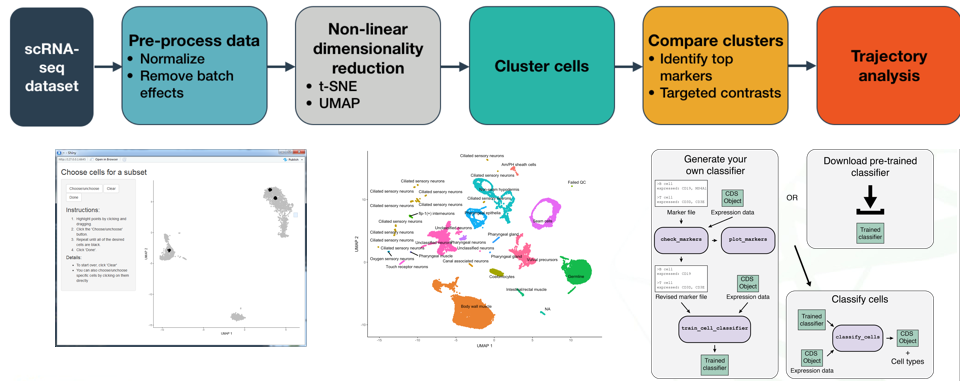
Monocel对象的生成
Monocle的cds对象其实在一定程度上非常类似于Seurat,只不过表示方法不一样,所以,我们可以很容易的从Seurat中取出需要的数据载入Monocle3
具体方法可以参考简书:scRNA-seq数据分析 || Monocle3
但是,monocle软件有自己的一套流程,囊括了标准化,归一化,降维,聚类等等,所以一般来说,我们都需要提供原始的未经处理的表达矩阵,但是,由于我们是整合了多样本的结果,而我们又不想使用monocle的批次效应去除方法,我们该怎么导入呢?
方法就是我们导入Seurat多样本整合并标准化的结果矩阵,然后,在后面的预处理过程中,取消掉标准化。
library(Seurat)library(monocle3)endo<-readRDS("DC.rds")DefaultAssay(endo)<-"integrated"data <- endo@assays$integrated@datapd <- endo@meta.data#the metadata have many rubbish info,we delete itnew_pd<-select(pd,stim,nCount_RNA,nFeature_RNA,percent.mt)new_pd$Cell.type<-Idents(endo)head(new_pd)stim nCount_RNA nFeature_RNA percent.mtAAACCTGGTATCAGTC-1_1 CTRL 4835 1383 3.536711AACCGCGCATTCCTGC-1_1 CTRL 4029 1230 2.655746AACGTTGAGCCCAATT-1_1 CTRL 2576 918 3.377329AAGACCTGTGGTTTCA-1_1 CTRL 2841 1044 5.455825AAGTCTGCATGAAGTA-1_1 CTRL 3731 1213 3.886358ACAGCCGCATCGGACC-1_1 CTRL 5915 1918 3.043111Cell.typeAAACCTGGTATCAGTC-1_1 DC2AACCGCGCATTCCTGC-1_1 MDSCAACGTTGAGCCCAATT-1_1 MDSCAAGACCTGTGGTTTCA-1_1 Dendritic.cells.activatedAAGTCTGCATGAAGTA-1_1 MDSCACAGCCGCATCGGACC-1_1 Dendritic.cells.activatedfData <- data.frame(gene_short_name = row.names(data), row.names = row.names(data))#create new cds objcds <- new_cell_data_set(data,cell_metadata = new_pd,gene_metadata = fData)
预处理
数据scale并采用PCA降纬
#we use normalized data,so we do not normalize itcds <- preprocess_cds(cds, num_dim = 30,norm_method = "none")
如果你非要使用Monocle的批次效应功能,可以尝试,batch列必须在你的pData里面
cds <- align_cds(cds, alignment_group = "batch")
采用umap的方法运行非线性降维
#umapcds <- reduce_dimension(cds,umap.n_neighbors = 20L)#color by seurat clusterplot_cells(cds,label_groups_by_cluster=FALSE,color_cells_by = "cell.type")
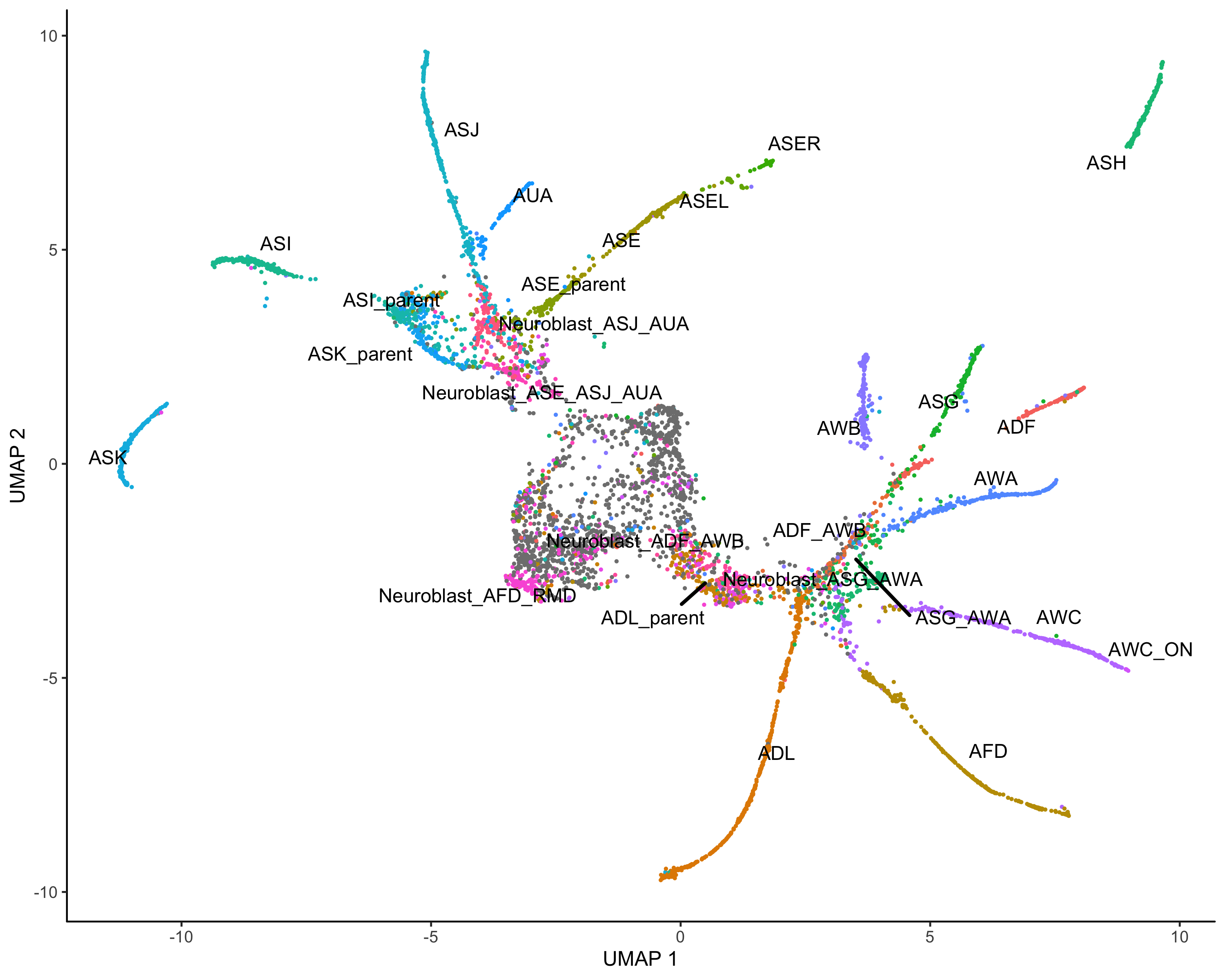
细胞聚类
#clustercds <- cluster_cells(cds,resolution = 0.5)#color by monocle clusterplot_cells(cds, color_cells_by = "partition",label_groups_by_cluster=FALSE)
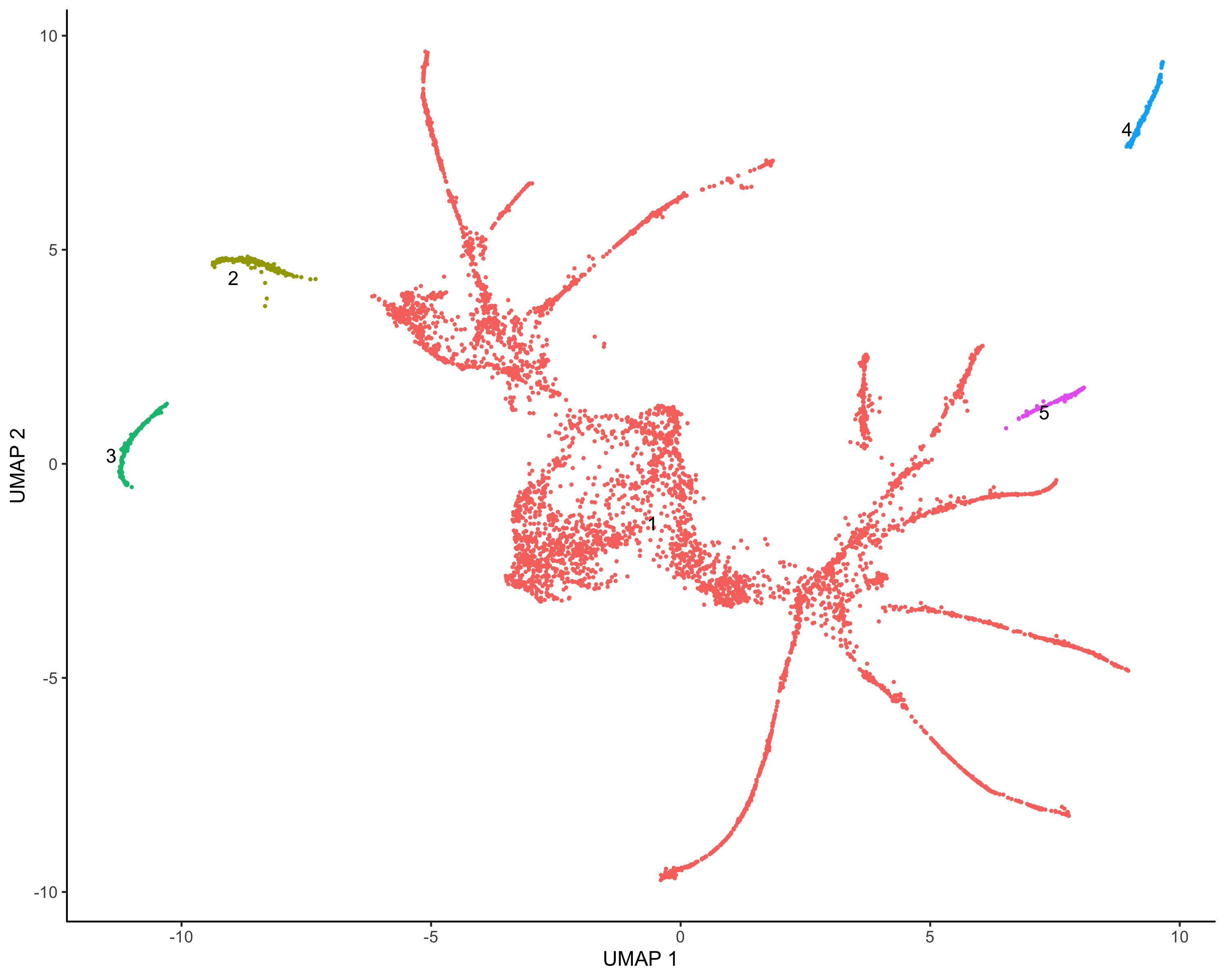
伪时间构建
其实说白了,就是基于图形以及表达量的变化关系,构建一个个基于起始表达的进化线或者说是分支,这里面肯定既有节点又有分枝。
cds <- learn_graph(cds)
画图展示:
plot_cells(cds,color_cells_by = "Cell.type",label_groups_by_cluster=FALSE,label_leaves=TRUE,label_branch_points=TRUE)
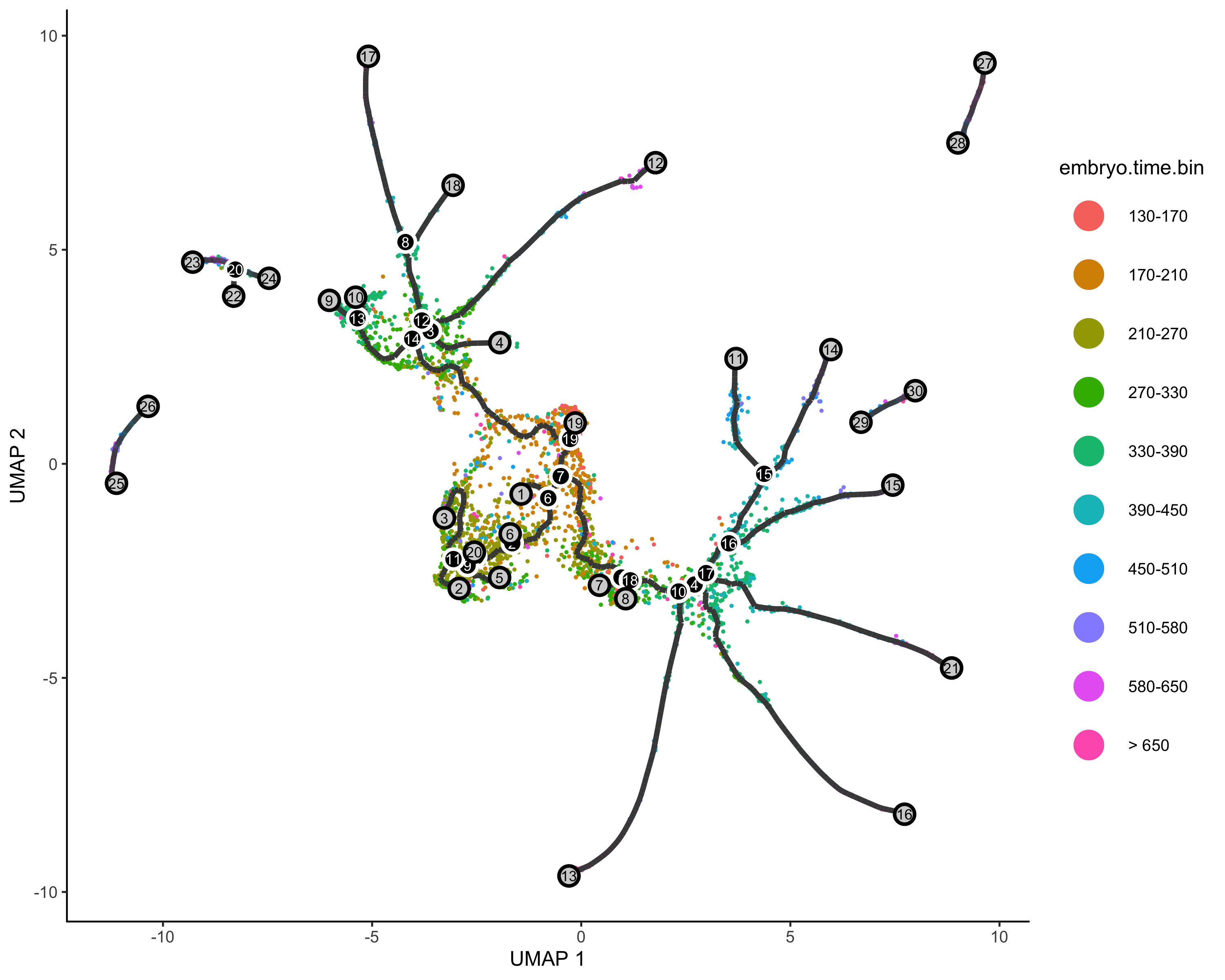
定义起始节点&&伪时间分析
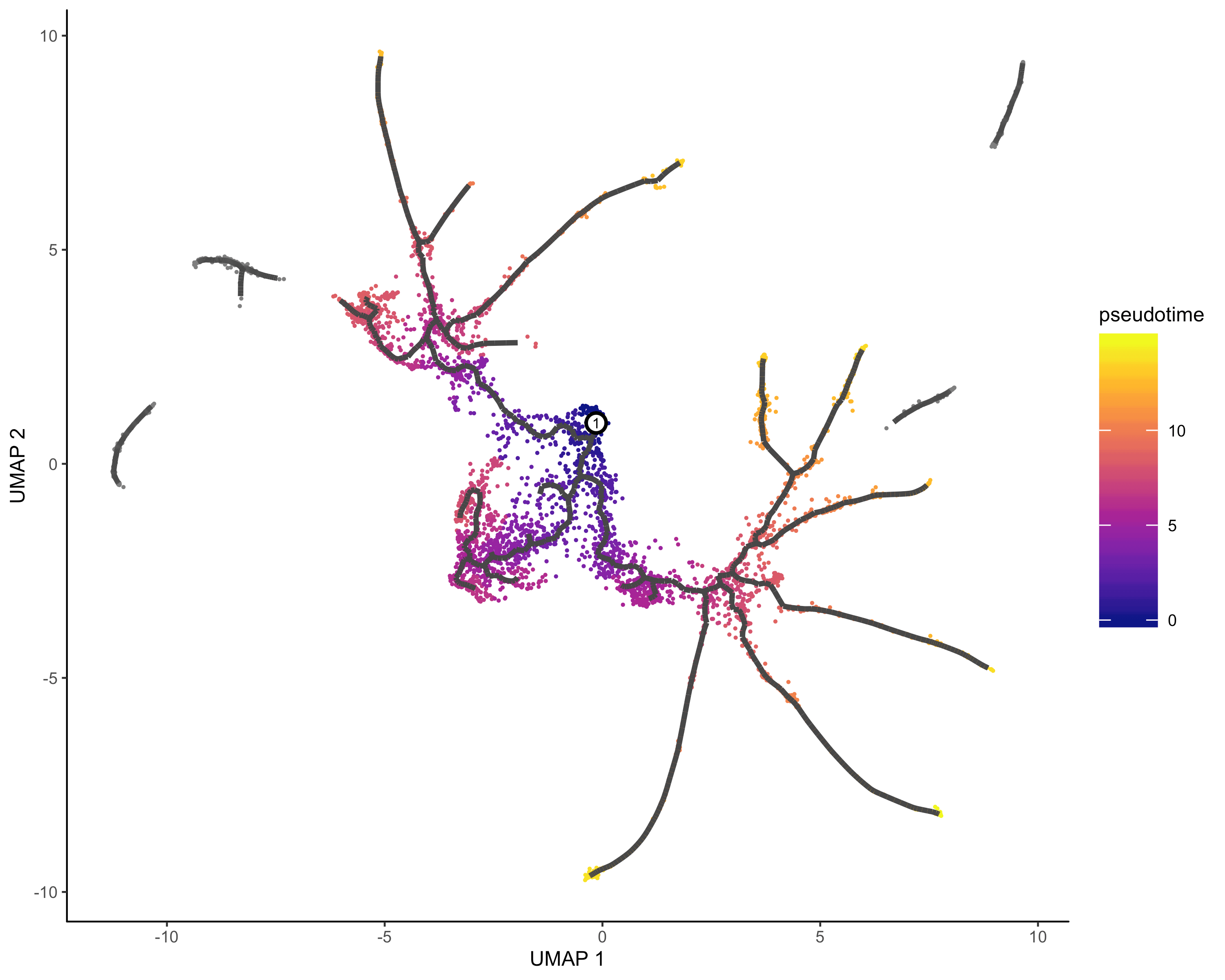
起始节点可以基于现有的知识,比如你已经将细胞类型注释好了,其中某种细胞就是起始的早期细胞,我们就可以把它作为root,如果不知道,我们只能使用1在的细胞群作为起始了
一个有用的定义根节点的函数
get_earliest_principal_node <- function(cds, time_bin="Dendritic.cells.activated"){cell_ids <- which(colData(cds)[, "Cell.type"] == time_bin)closest_vertex <-cds@principal_graph_aux[["UMAP"]]$pr_graph_cell_proj_closest_vertexclosest_vertex <- as.matrix(closest_vertex[colnames(cds), ])root_pr_nodes <-igraph::V(principal_graph(cds)[["UMAP"]])$name[as.numeric(names(which.max(table(closest_vertex[cell_ids,]))))]root_pr_nodes}
定义根节点
#order cellcds <- order_cells(cds, root_pr_nodes=get_earliest_principal_node(cds))#pseudotime analysisplot_cells(cds,color_cells_by = "pseudotime",label_cell_groups=FALSE,label_leaves=FALSE,label_branch_points=FALSE,graph_label_size=1.5)
在基因层面探索伪时间
寻找随时间变异的基因
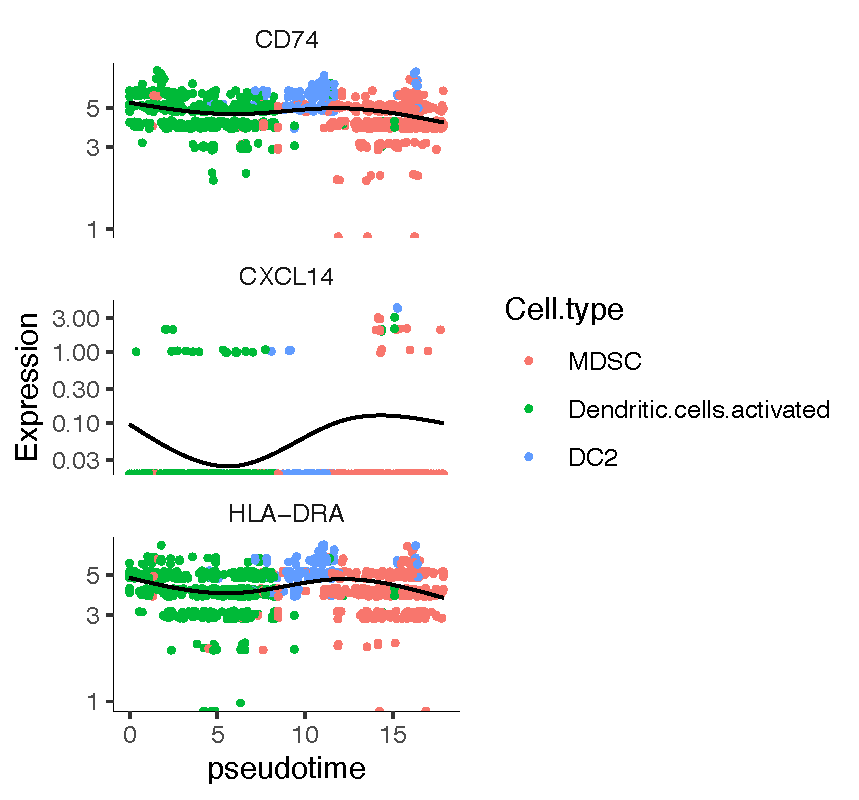
diff_gene <- graph_test(cds, neighbor_graph="principal_graph", cores=4)id <- row.names(subset(diff_gene, q_value < 0.05))head(id)[1] "CD74" "CXCL14" "HLA-DRA" "GAST" "C1QB" "HBA1"
在cds对象中寻找基因并作图,为了看出变化,可以选择case组中的表达量
CASE_genes <- c("CD74", "CXCL14", "HLA-DRA")CASE_lineage_cds <- cds[rowData(cds)$gene_short_name %in% AFD_genes,colData(cds)$stim %in% c("CASE")]#plot genesplot_genes_in_pseudotime(AFD_lineage_cds,color_cells_by="Cell.type")
教程到此结束,原创禁止转载
文中图片大部分取自官网,仅作为示例。

若有收获,就点个赞吧
0 人点赞
