前言
CRISPR 基因编辑技术近年来大热,因具有精确的编辑和较高的效率,成为动植物基因改造的热门技术,传统技术的分析过程采用 sanger 测序方法,通过对测序峰图的分析对其插入和删除效率进行分析,而随着二代测序技术逐渐成熟,结合 CRISPR 和 NGS 测序的数据分析必将大热,本文将总结一些 NGS 数据分析的软件予以参考
软件
1.CRISPResso2
CRISPresso2 软件是老牌的 crispr 数据分析软件,其功能多样,但是分析速度有待提高。
1.1 原理
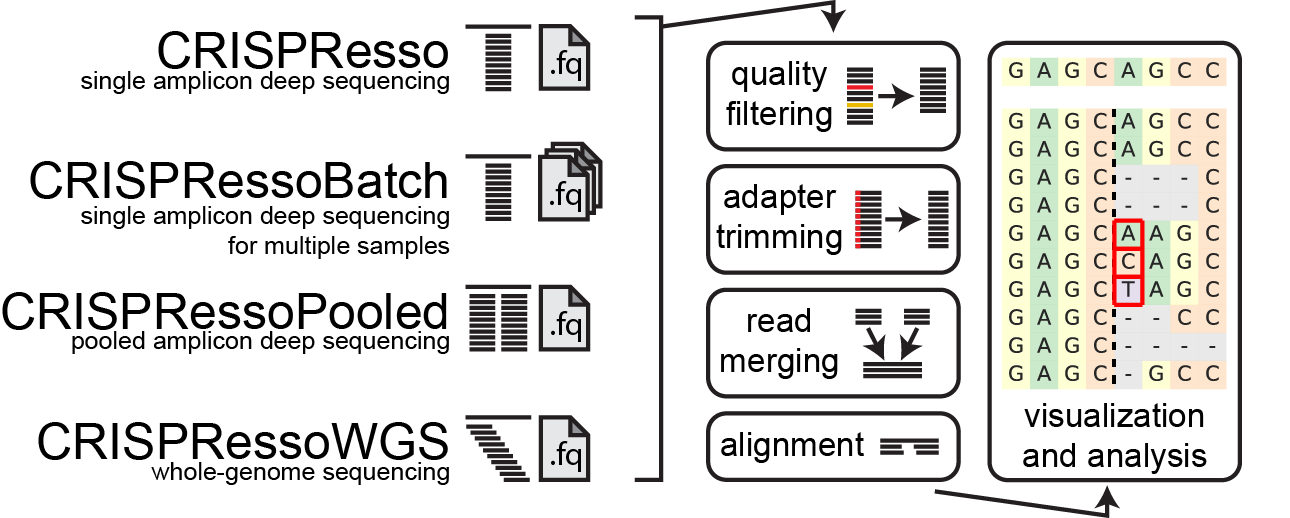
该软件通过对原始序列进行拼接(FLASH),然后运用前文所讲的全局比对的方式进行序列 alan,对结果进行统计并作图
The CRISPResso suite accommodates single or pooled amplicon deep sequencing, WGS datasets and allows the direct comparison of individual experiments. In fact four additional utilities are provided:
- CRISPRessoPooled: a tool for the analysis of pooled amplicon experiments
- CRISPRessoWGS: a tool for the analysis of WGS data or prealigned reads in .bam format
- CRISPRessoCompare:a tool for the comparison of two CRISPResso analyses, useful for example to compare treated and untreated samples or to compare different experimental conditions
- CRISPRessoPooledCompare: a tool to compare experiments involving several regions analyzed by either CRISPRessoPooled or CRISPRessoWGS
简单说,包含很多功能模块,pool 是进行大批量数据和同一个参考基因组进行比对,同时返回每个数据组数据的程序;WGS 则是与全基因组进行比对的程序;Compare 则是带有 CK 对照组数据进行对比分析的程序,由此可见,该程序的复杂度很高,功能相对完善。
1.2 安装方法
建议通过conda的方式来安装.
打开终端并键入以下命令:
conda config --add channels defaultsconda config --add channels biocondaconda config --add channels conda-forge
为了避免安装对原有环境的影响,建议添加额外的环境
conda create -n crispresso2_env -c bioconda crispresso2 python=2.7
安装完成后,可以通过执行以下命令验证安装是否完好:
CRISPResso -h
1.3 使用方法
因为二代测序多为双端测序,因此,这里只简单的介绍双端测序的使用方法
CRISPResso -r1 reads1.fastq.gz -r2 reads2.fastq.gz -a ref.fa -g gRNA_no_PAM
2. CRISPR-DAV
CRISPR-DAV也是一个 pipline,不过是用 perl 语言书写的,本人对 perl 语言不甚了解,所以就不过多解释了,但是 git 上作者推荐使用 docker 安装,因此,感兴趣的可以尝试一波
2.1 安装方式
3.Cas-analyzer
Cas-Analyzer 是一个快速的在线分析软件,其程序基于 JS 语言在网页上进行计算,其计算速度飞快,准确率也很高,但是可惜的是,没有原始数据下载,也没有本地版本
4.CRISPRMatch
CRISPRMatch同样是基于 Python 程序的 pipline,作者在文章中介绍说软件相比于传统的软件具有很大的优势,其中之一便是安装和使用简单,但实际体验一般
4.1 安装方法
4.1.1 安装 anaconda
$ yum install wget git$ mkdir /home/software$ cd /home/software$ wget https://repo.continuum.io/archive/Anaconda3-5.0.1-Linux-x86_64.sh$ bash Anaconda3-5.0.1-Linux-x86_64.sh
4.1.2 安装依赖
$ conda install bwa \samtools \picard \flash \matplotlib \pysam \pandas \argparse \numpy \
4.1.3 下载软件并测试
$ cd /home/software$ git clone https://github.com/zhangtaolab/CRISPRMatch.git$ python3 /home/software/CRISPRMatch/CRISPRMatch.py -husage: CRISPRMatch [-h] [--version] [-b BWA] [-sm SAMTOOLS] [-pi PICARD] -gGENOME -i INPUT -gi GROUPINFO [-s SAVED] [-r RESULT][-t THREADS] [--docker DOCKER]CRISPRMatch is for location findingoptional arguments:-h, --help show this help message and exit--version show program's version number and exit-b BWA, --bwa BWA bwa path-sm SAMTOOLS, --samtools SAMTOOLSsamtools path-pi PICARD, --picard PICARDpicard path-g GENOME, --genome GENOMEfasta format genome file-i INPUT, --input INPUTsample information input file-gi GROUPINFO, --groupinfo GROUPINFOgroup information input file-s SAVED, --save SAVEDtmp saved folder-r RESULT, --result RESULTresult saved folder-t THREADS, --threads THREADSthreads number or how may cpu you wanna use
4.2 使用方法
软件使用需要提前转备好 3 个文件,分别是
- 参考基因序列
- 样本信息
- 分组信息
具体的格式应该严格参照作者提供的示例数据格式(sampledata,sampledata2)
3.4 运行
$ cd /home/software/CRISPRMatch/$ python3 CRISPRMatch_paired.py -g sampledata2/Samples_gene.fa -i sampledata2/sample_infor.txt -gi sampledata2/group_info.txt -t 2

若有收获,就点个赞吧
0 人点赞
