以下的突变检测都是基于NGS测序的结果,但是需要预先使用bwa等工具预先比对到参考序列或者基因组上,
然后根据要研究的目标区域提取比对上的序列作进一步的分析,一个有用的方式就是使用samtools.
samtools view -b -F chrN:NNNNN-NNNNN > contig_1.bam
因为提取出来的都是短序列,因此,我们可以在下游使用全局比对(Needleman-Wunsch)来获取比对详情。
经过简单的处理,我将在bam文件中提取比对到参考序列上的短序列并存入Fasta格式
query:
>10613CATTGAGATGGTGTGGGAAGGGGCCCCATTGAGATAGTGTGGCCCGTTTGAAGGGGCCCCATTGAGATAGAAGGGGCCCCATTGAGATAGAAGGGGCCCCATTGAGATAGTGTGGCATTGAGATAGGCCCCATTGAGATAGTGTGGGCCCCATTGAGATAGTGTGGGAAGGGGCCCCATTGAGATAGTGTGGGAAGGGGCCCCATTGAGATAGTGTGGGCCCCATTGAGATAGTGTGGGAAGGGGCCCCATTGAGATAGTGTGGGAAGGGGCCCCATTGAGATAGTGTGGGCCCCATTGAGATAGTGCCCCATTGGGCCCCATTGAGATAGGGCCCCATTGAGATATGTGGGGAAGGGGCCC'''''
ref:
CATTGAGATAGTGTGGGGAAGGGGCCC
突变检测
目标:统计出indel,snp等常见突变的位置,突变量(nbp突变)以及突变频率
需要借助python多序列比对模块,这里我们采用github上的一个
##导入所有需要的模块from align.calign import aligner as calignerfrom align.matrix import DNAFULL, BLOSUM62from collections import Counterimport refrom pyfaidx import Fastaimport pandas as pdimport numpy as np##定义读入fasta序列的函数def read_res(fs):fasta={}with open(fs,'r') as f:for line in f:if line.startswith(">"):seq_lst=[]name=line.strip("\n").replace(">","")else:seq_lst.append(line.strip("\n"))fasta[name]=seq_lstreturn fasta###采用正则表达式来提取突变def clean_insertion(ref,query):ins_lst=[]delet=[]for site in re.finditer(r"-+",ref):(start,end)=site.span()length=end-startins_lst.append("{}I{}".format(start+1,length))delet.append(list(range(start,end)))delet_2=[y for x in delet for y in x]query="".join([query[i] for i in range(len(query)) if i not in delet_2])ref=ref.replace("-","")ins_lst2=";".join(ins_lst)return (ref,query,ins_lst2)def detective_del(ref,query):deletion=[]for st in re.finditer(r"-+",query):(start,end)=st.span()length=end-startdeletion.append("{}D{}".format(start+1,length))deletion2=";".join(deletion)return (ref,query,deletion2)def detective_snp(ref,query):snp_lst=["{}M1".format(st+1) for st in range(len(ref)) if (query[st]!=ref[st])&(query[st]!="-")&(ref[st]!="-")]snp=";".join(snp_lst)return (ref,query,snp)def crispr_det(querys,ref):query_lst=Counter(querys)alreads=sum(list(query_lst.values()))fin=[]for key,val in query_lst.items():bijiao=caligner(ref,key,method="global",matrix=DNAFULL)(ref,query,insertions)=clean_insertion(bijiao[0].seq1.decode(),bijiao[0].seq2.decode())(ref,query,deletions)=detective_del(ref,query)(ref,query,mutations)=detective_snp(ref,query)tmp={"reference":ref,"query":query,"ins":insertions,"del":deletions,"mut":mutations,"percentage":"{:.2f}".format(val/alreads*100)}fin.append(tmp)return fin##读入query&reftest=read_res("query.fa")reference = Fasta("reference.fasta")querys=test['10613']ref=reference["10613"][113:140].seq ## 请查阅 pyfaidx 模块介绍df=pd.DataFrame(crispr_det(querys,ref))df['percentage']=df['percentage'].astype(np.float64)df=df.sort_values('percentage',axis=0,ascending=False)df.head(10)
运行完后可以查看最终获得的结果:
| reference | query | ins | del | mut | percentage | |
|---|---|---|---|---|---|---|
| 6 | CATTGAGATAGTGTGGGGAAGGGGCCC | CATTGAGATAGTGTGGG-AAGGGGCCC | 18D1 | 16.53 | ||
| 5 | CATTGAGATAGTGTGGGGAAGGGGCCC | CATTGAGATAGTGTGGG———-CCC | 18D7 | 10.93 | ||
| 10 | CATTGAGATAGTGTGGGGAAGGGGCCC | CATTGAGATA-TGTGGGGAAGGGGCCC | 11D1 | 10.10 | ||
| 30 | CATTGAGATAGTGTGGGGAAGGGGCCC | CATTGAGATAGTGTGGGG———CCC | 19D6 | 7.60 | ||
| 2 | CATTGAGATAGTGTGGGGAAGGGGCCC | CATTGAGATAG———-AAGGGGCCC | 12D7 | 5.34 | ||
| 14 | CATTGAGATAGTGTGGGGAAGGGGCCC | CATTGAGATAGTGTGG————CCC | 17D8 | 4.42 | ||
| 3 | CATTGAGATAGTGTGGGGAAGGGGCCC | CATTGAGATAGTGTGG—————- | 17D11 | 3.67 | ||
| 9 | CATTGAGATAGTGTGGGGAAGGGGCCC | CATTGAGATAG-G-G————-CCC | 12D1;14D1;16D9 | 3.09 | ||
| 13 | CATTGAGATAGTGTGGGGAAGGGGCCC | CATTGAGA-AG-G-GG————CCC | 9D1;12D1;14D1;17D8 | 2.92 | ||
| 18 | CATTGAGATAGTGTGGGGAAGGGGCCC | C—-GA-A—G-G-GG————CCC | 2D3;7D1;9D2;12D1;14D1;17D8 | 2.67 |
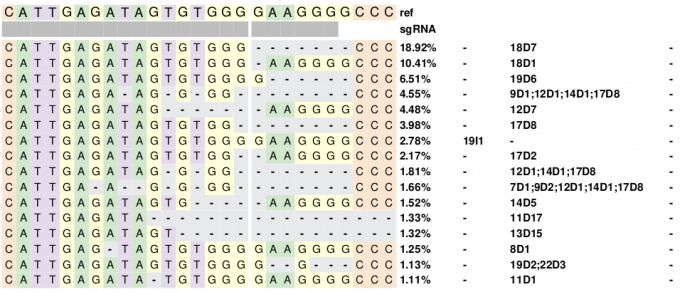
对以上结果的可视化展示
因为R语言画图是很方便的,所以这里采用R语言热图展示突变情况
##数据处理data<-read_delim("ontarget6.mutation.tsv",delim = "\t")ref<-stringr::str_split_fixed(data$reference[1],"",n=27)ref<-as.character(ref)need<-data%>%filter(percentage>=1)%>%tidyr::replace_na(replace = list(ins="-",del="-",mut="-"))plotData<-stringr::str_split_fixed(need$query,"",n=27)gRNA<-c(rep("1",23),rep("2",4))##画Heatmap(plotData,cluster_rows = FALSE,cluster_columns = FALSE,column_split = c(rep("1",17),rep("2",10)),column_title = NULL,col = c("A"="#d1e8cd","G"="#ffffda","C"="#fae6d1","T"="#e4daec","-"="#e8e9ea"),cell_fun = function(j,i,x,y,wt,ht,fill){if(plotData[i,j]!=ref[j]){grid.text(sprintf("%s", plotData[i, j]), x, y, gp = gpar(fontsize = 10,fontface="bold"))}else{grid.text(sprintf("%s", plotData[i, j]), x, y, gp = gpar(fontsize = 10))}},top_annotation = HeatmapAnnotation(ref=anno_simple(ref,pch=ref,col = c("A"="#d1e8cd","G"="#ffffda","C"="#fae6d1","T"="#e4daec")),sgRNA=anno_simple(gRNA,col = c("1"="grey","2"="white")),annotation_name_gp = gpar(fontsize=10,fontface="bold")),right_annotation = rowAnnotation(percentage=anno_text(sprintf("%s%%",need$percentage),gp=gpar(fontsize=10,fontface="bold")),insertion=anno_text(need$ins,gp=gpar(fontsize=10,fontface="bold")),deletion=anno_text(need$del,gp=gpar(fontsize=10,fontface="bold")),substitutions=anno_text(need$mut,gp=gpar(fontsize=10,fontface="bold")),gap=unit(2,'char')),show_heatmap_legend = FALSE)
结果

若有收获,就点个赞吧
0 人点赞
