eccDNA 概述
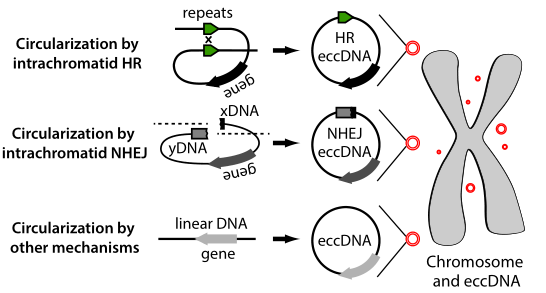
环状DNA是自然界普遍存在的一种DNA分子形式,例如细菌或酵母等微生物的基因组DNA、细菌质粒、线粒体DNA (mtDNA) 等等都是环状DNA分子。真核生物中还有一类特殊的环状DNA分子,它们是从正常基因组中分离或脱落下来,游离于染色体基因组之外,以特殊的方式参与生理或病理过程。由于它们是在染色体之外独立存在的DNA分子,因此统称为染色体外DNA,又常常是环状的,因此称其为染色体外环状DNA (extrachromosomal circular DNA, eccDNA)。染色体外DNA在很多物种中均存在,包括酵母、线虫、果蝇、哺乳动物、植物等等[1]。eccDNA往往在肿瘤和衰老过程中富集,以特殊的方式参与肿瘤和衰老的发生发展进程[1],因此备受关注。
基于NGS测序的eccDNA研究进展
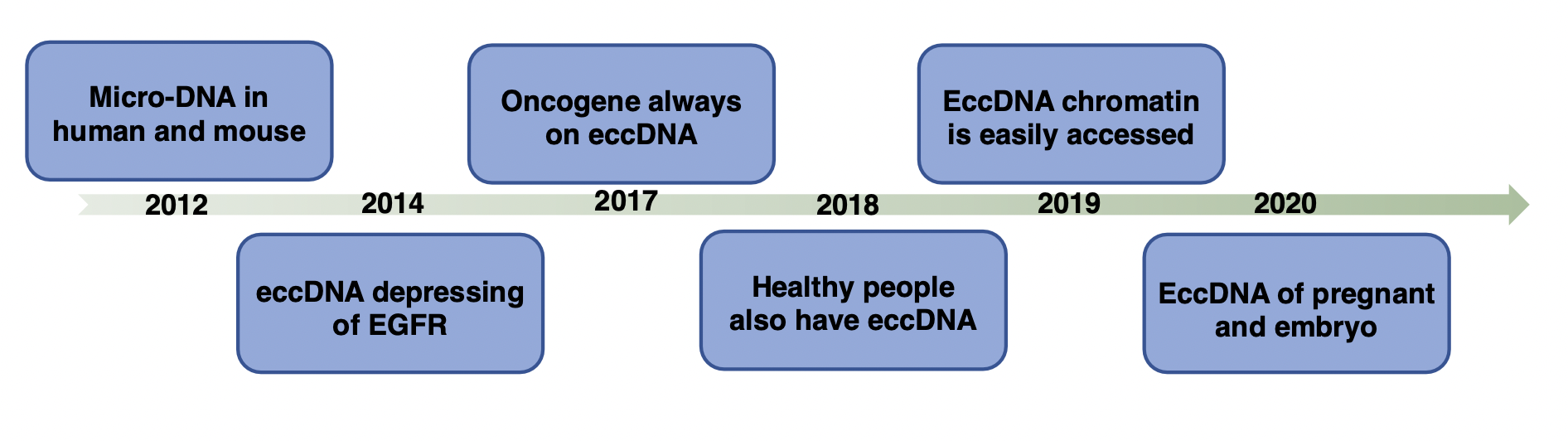
人类基因组计划的完成和二代测序技术的兴起带动了eccDNA的发展。2012年,Shibata, Y.等报道从小鼠和人的细胞中发现了一类小的染色体外环状DNA分子,它们大小在200-400bp不等的染色体外环状DNA,但这些DNA分子与上述的eccDNA并不一致,他们称其为microDNA[2]。2014年,Paul S. Mischel团队发现胶质瘤细胞中存在携带EGFRvIII突变型基因的eccDNA,介导了EGFR抑制剂耐药[3]。2017年,Paul S. Mischel团队对17种肿瘤的2572种细胞系的全基因组测序 (WGS) 分析,发现超过一半的人类肿瘤中存在eccDNA,这些eccDNA分子经常携带肿瘤驱动基因[4]。2018年,Moller, H. D.等报道从健康人的肌肉和血液细胞中分离到超过十万种的eccDNA分子,它们绝大部分都携带基因或基因的片段,长度往往小于25kb[5]。这一报道表明eccDNA可能普遍存在。2019年,Paul S. Mischel团队报道eccDNA存在显著的染色体开放状态和更远距离相互作用的形式,提高了对eccDNA的作用机制认识[6]。同期,Jeremy N. Rich和Peter C. Scacheri等对eccDNA中染色质结构和增强子状态进行了分析,表明eccDNA中非编码区的增强子功能也在eccDNA所携带的癌基因表达调控中发挥功能,也进一步表明了eccDNA特殊的染色质结构及其功能[7]。最近,香港中文大学卢煜明(Dennis Lo)教授领导的研究团队,近日从孕妇血浆中鉴定出母亲和胎儿的环状游离DNA,为无创产前检测提供了一种新型的生物标志物,同时也提供了分离孕妇和胎儿eccDNA的生物信息学方法[8]。
已有的NGS分析方法和流程
AmpliconArchitect (AA)[9]
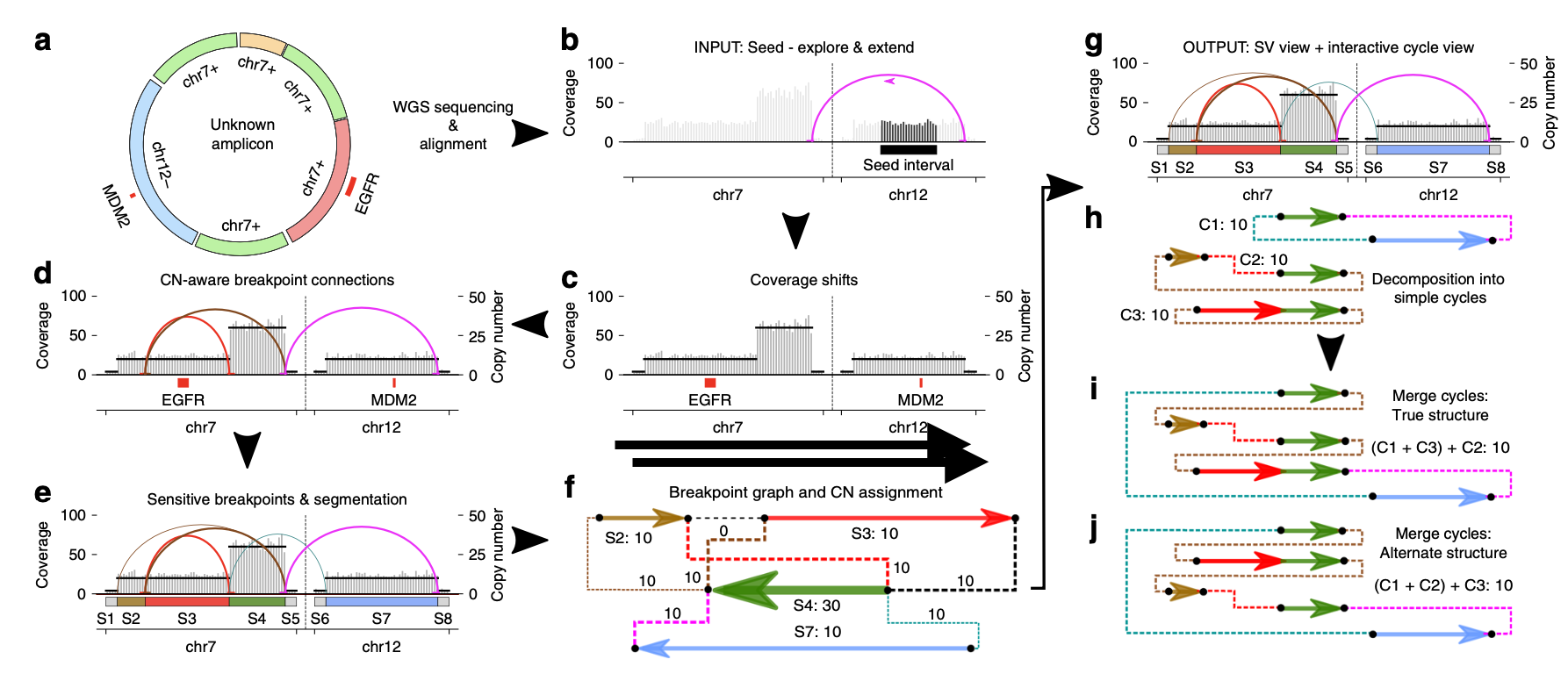
AmpliconArchitect在算法的每一步都会生成有用的输出(详细信息如下):
AmpliconArchitect算法
AA是一个完整的生物信息分析流程,它使用以下步骤来预测扩增子的结构:
- 种子间隔区确定:确定要重建的每个扩增子的间隔列表。
- SV检测:使用每个扩增子中的覆盖范围和不一致的读对来检测拷贝数变化和结构变异。
- 断点图构造:构造一个断点图,该断点图由序列边(基因组片段),断点边(连接的基因组位置对)和源顶点(可选)组成,并预测所有边的拷贝数。
- 循环分解:将断点图分解为简单的循环,以简单的方式表示预测的扩增子结构。
- 交互式循环合并:提供一个Web界面,以交互方式合并和修改循环以探索候选结构。
AmpliconArchitect的用途
AA可以在WGS数据中发现潜在的eccDNA片段,并通过片段和端点图组成多种结构,并尽可能还原真实的eccDNA结构。因为AA软件的原理是基于eccDNA的两个特征:1)拷贝数显著扩增,2)是一种特殊的环状结构突变(SV)。 因此,理论上,ATAC-seq,CIRCLE-seq的数据同样可以使用AA来分析。
可用的AA程序已经被封装为docker,使用说明可以在github地址获取 (https://github.com/virajbdeshpande/AmpliconArchitect)
Circle_finder[10]
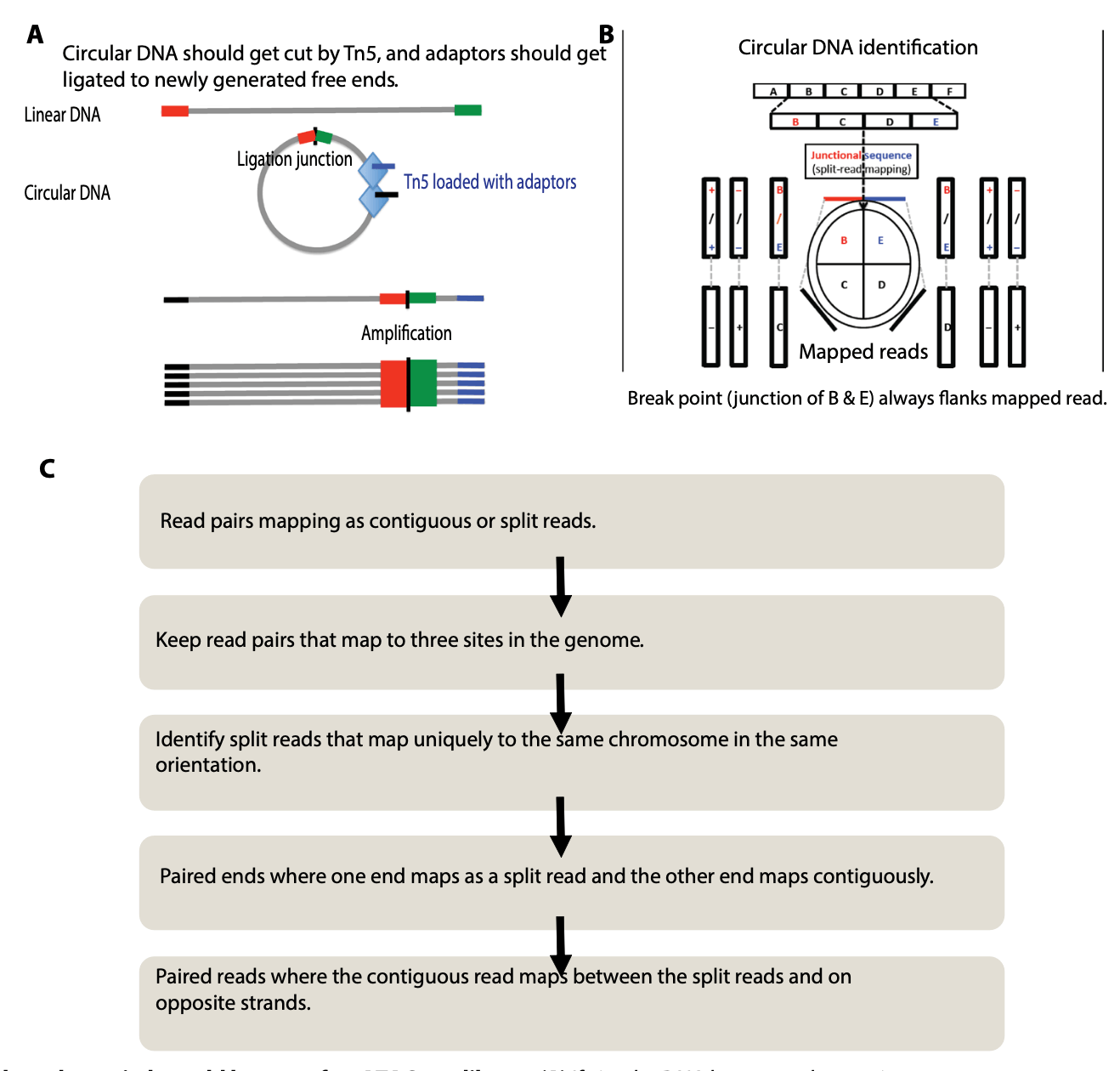
Circle_finder算法和用途
Circle_finder使用现有的NGS数据分析软件,通过shell脚本串联在一起,可以分析ATAC-seq数据中的eccDNA片段。包括使用samblaster软件区分不连续比对和拆分比对,使用bedtools来处理基因组区间问题等。
Circle_finder同样可以用于WGS数据分析,但是无论是哪种数据,官方都推荐可以对环状DNA进行生物学富集。可用的脚本可以通过github链接获取(https://github.com/pk7zuva/Circle_finder/blob/master/circle_finder-pipeline-bwa-mem-samblaster.sh)
Circle-Map[11]
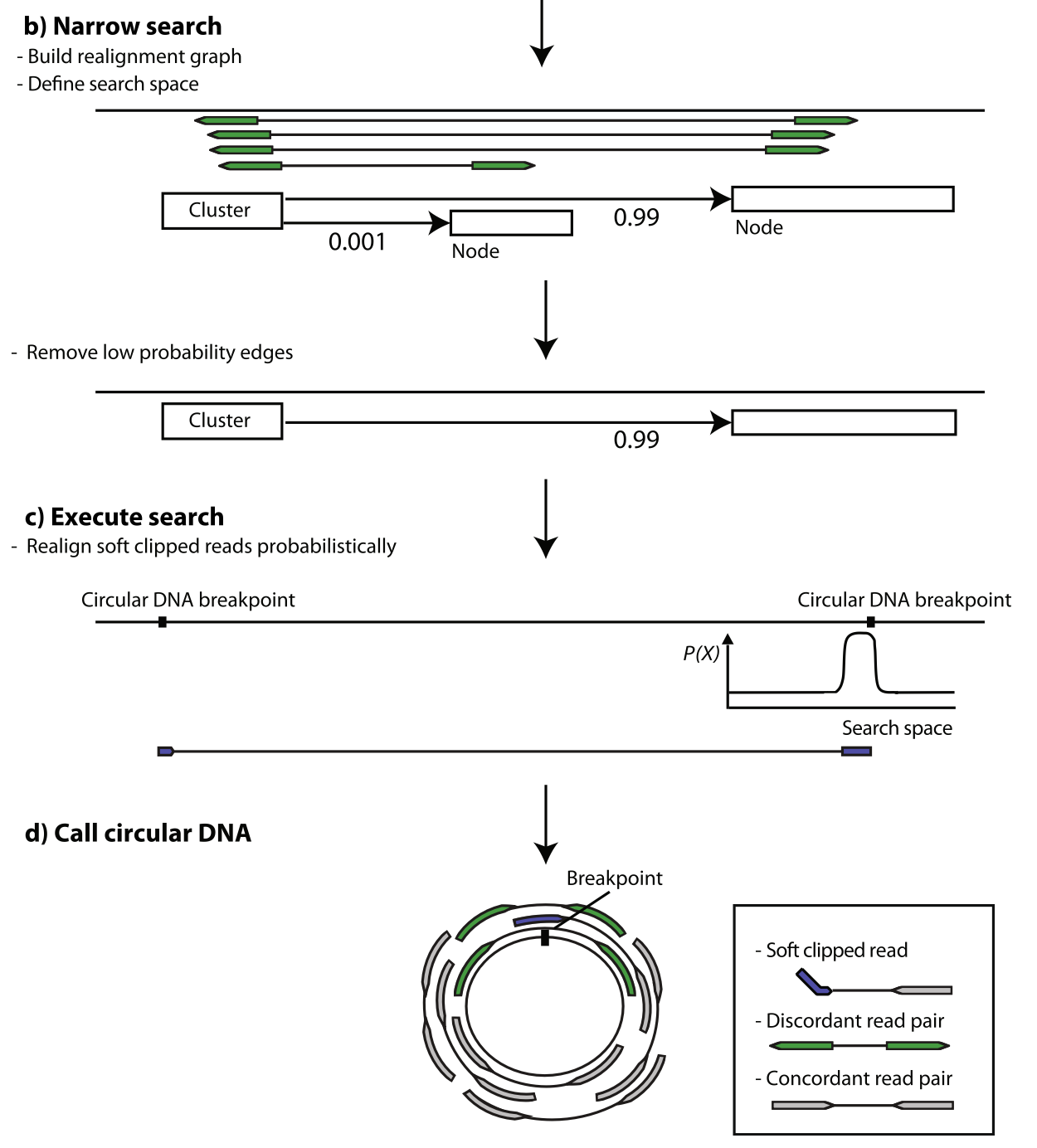
Circle-Map算法和用途
Circle-Map将读长序列与参考基因组的比对 (例如,由BWA-MEM生成的BAM文件) 作为输入,并且像其他方法一样,它将使用这些比对来检测将读取分为两部分的情况 (拆分读长) 检测支持环状DNA结构的基因组重排。
但是,这种方法会导致许多拆分读长对齐方式丢失,因为比对软件无法映射读长的两个拆分段,要么是因为它们太短,要么是因为它们对齐到了太多地方。在这种情况下,比对软件将报告包含某些未映射碱基的读长对齐 (软剪切)。
与其他方法不同,Circle-Map能够概率性地将未映射的部分重新比对重排为环状DNA端点图,从而正确比对软剪切读长的两个片段,这样可以更精确地检测环状DNA断裂点。
可用的Circle-Map软件可以通过github链接获取,安装也很方便,基于bio-conda (https://github.com/iprada/Circle-Map)。 Circle-Map仅适用于CIRCLE-seq数据 (线性DNA被线性消除后的数据)
参考文献
Barreto, S.C., M. Uppalapati, and A. Ray, Small Circular DNAs in Human Pathology. Malays J Med Sci, 2014. 21(3): p. 4-18.
Shibata, Y., et al., Extrachromosomal microDNAs and chromosomal microdeletions in normal tissues. Science, 2012. 336(6077): p. 82-6.
Nathanson, D.A., et al., Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science, 2014. 343(6166): p. 72-6.
Turner, K.M., et al., Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature, 2017. 543(7643): p. 122-125.
Moller, H.D., et al., Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat Commun, 2018. 9(1): p. 1069.
Wu, S., et al., Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature, 2019.
Morton, A.R., et al., Functional Enhancers Shape Extrachromosomal Oncogene Amplifications. Cell, 2019.
Sarah T. K. Sin, Peiyong Jiang, Jiaen Deng, et.al. Identification and characterization of extrachromosomal circular DNA in maternal plasma. PNAS January 3, 2020
Deshpande, V. et al. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat. Commun. 10, 392 2019
Kumar P, Dillon LW, Shibata Y, Jazaeri AA, Jones DR, Dutta A. Normal and Cancerous Tissues Release Extrachromosomal Circular DNA (eccDNA) into the Circulation. Mol. Cancer Res. Sep; 15(9): 1197-1205, 2017.
Prada-Luengo, I., Krogh, A., Maretty, L. et al. Sensitive detection of circular DNAs at single-nucleotide resolution using guided realignment of partially aligned reads. BMC Bioinformatics 20, 663 2019

若有收获,就点个赞吧
0 人点赞
