WGCNA的代码有点复杂,大概分成 确定软阈值、模块与性状相关性热图、挑感兴趣模块进一步分析、TOM矩阵做基因相关性热图等,我是觉得最后一个图没啥意思,什么都看不出来,而且对电脑设备要求也高,所以一般只做前两步,挑出和性状相关性比较强的模块内基因,做下一步分析。
R包和数据导入
exp为表达矩阵,cl为临床信息
library(WGCNA)load('Rdata/exp_cl.Rdata')
输入数据准备
WGCNA的数据需要log2,可以用fpkm或者tpm,我看有公众号说cpm也可以,因为cpm转换容易,就用了这个
关于跑多少基因:可以提取mad/变异系数/方差/blabla(看个人选用的标准了)前5k,也有人只做差异基因,我还看到有人做的是表达量前五千的基因,其实我也不确定这一步的筛选方法。因为我开局WGCNA了,所以就挑取了表达量前5k的基因
这一步是为了得到表达矩阵的输入矩阵 datExpr
exprSet <- log2(edgeR::cpm(exp)+1)exprSet <- t(exprSet) # WGCNA 是对基因进行聚类,需要转置
WGCNA的输入数据这里考虑到了离群值,需要筛选一下,众所周知,凡是涉及到筛选的都是玄学,所以就做个树状图看一下有没有问题就行
WGCNA不推荐打包跑,因为中间需要注意的地方有点多,也确实很费时间
sampleTree <- hclust(dist(exprSet), method = "average") # 生成聚类树sizeGrWindow(12,9)par(cex = 0.6)par(mar = c(0,4,2,0))plot(sampleTree, main = "Sample clustering to detect outliers", sub="", xlab="", cex.lab = 1.5, cex.axis = 1.5, cex.main = 2)
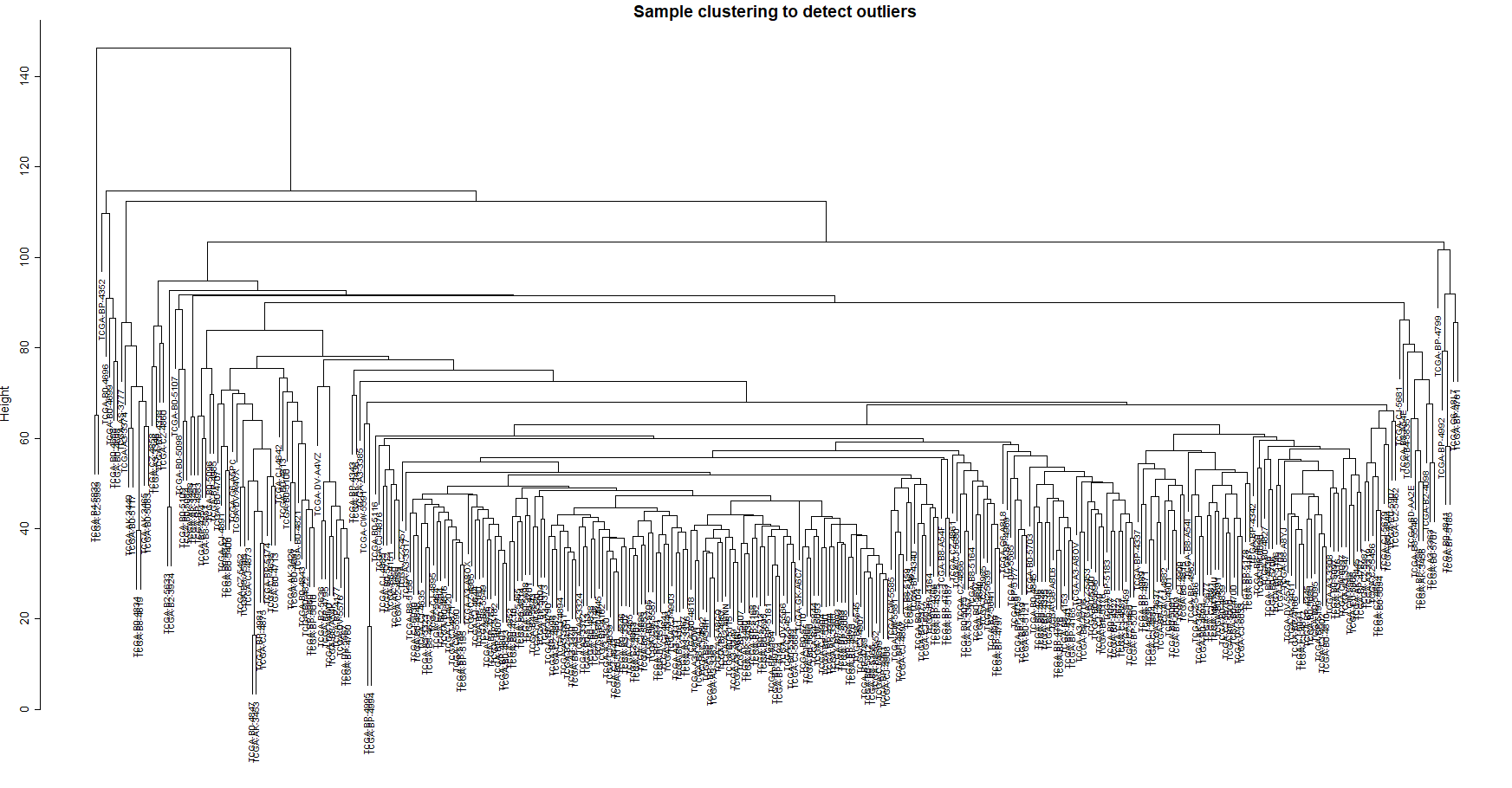
我没有去离群值,如果要去,代码可参考↓ (以200以上排除为例)
abline(h = 200, col = "red") # 有一说一,这个去离群sample有点玄学,全凭个人感觉clust <- cutreeStatic(sampleTree, cutHeight = 200, minSize = 0)keepSamples <- (clust==1)datExpr <- exprSet[keepSamples, ]dim(datExpr) # 输入矩阵okcollectGarbage() # 执行垃圾回收,不是很清楚这一步的目的sampleTree2 <- hclust(dist(datExpr), method = "average") # 生成新的聚类树
确定软阈值
确定软阈值的目的就是保证基因符合无尺度网络(scale-free network),无尺度网络可以理解为:20%的人掌握着80%的财富。
powers = c(c(1:10), seq(from = 12, to=20, by=2))sft = pickSoftThreshold(datExpr, powerVector = powers, verbose = 5) # 有点慢,分析软阈值的一个函数sizeGrWindow(9, 5)par(mfrow = c(1,2))cex1 = 0.9plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],xlab="Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",type="n",main = paste("Scale independence"));text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],labels=powers,col="red");abline(h=0.85,col="red")plot(sft$fitIndices[,1], sft$fitIndices[,5],xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n",main = paste("Mean connectivity"))text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers,col="red")# 看不出来的话 可以简单粗暴运行下面这句,WGCNA作者说,软阈值大概就在6的附近# power = sft$powerEstimatecor <- WGCNA::cor # 因为基础包里也有cor,可能会报错,先来这么一句,下面运行完再改回来net = blockwiseModules(datExpr,power = 6, # 软阈值 记得改TOMType = "unsigned", # positvie和negative都要,我看yt上的老师说,她倾向于signed,我分别跑了一下,感觉大体不差minModuleSize = 30, # 最小模块选择reassignThreshold = 0,mergeCutHeight = 0.25,numericLabels = TRUE,pamRespectsDendro = FALSE,saveTOMs = TRUE,saveTOMFileBase = "WGCNA_TOM",verbose = 3,deepSplit = 2)cor<-stats::corclass(net)names(net)table(net$colors)moduleLabels = net$colorsmoduleColors = labels2colors(net$colors) # 每个基因都赋予了一个模块颜色sizeGrWindow(12, 9)plotDendroAndColors(net$dendrograms[[1]], moduleColors[net$blockGenes[[1]]],"Module colors",dendroLabels = FALSE, hang = 0.03,addGuide = TRUE, guideHang = 0.05) #生成层次聚类树和模块对应图
sft$powerEstimate 直接用这个可能有点太粗暴了,一般结果还行,有时候不太好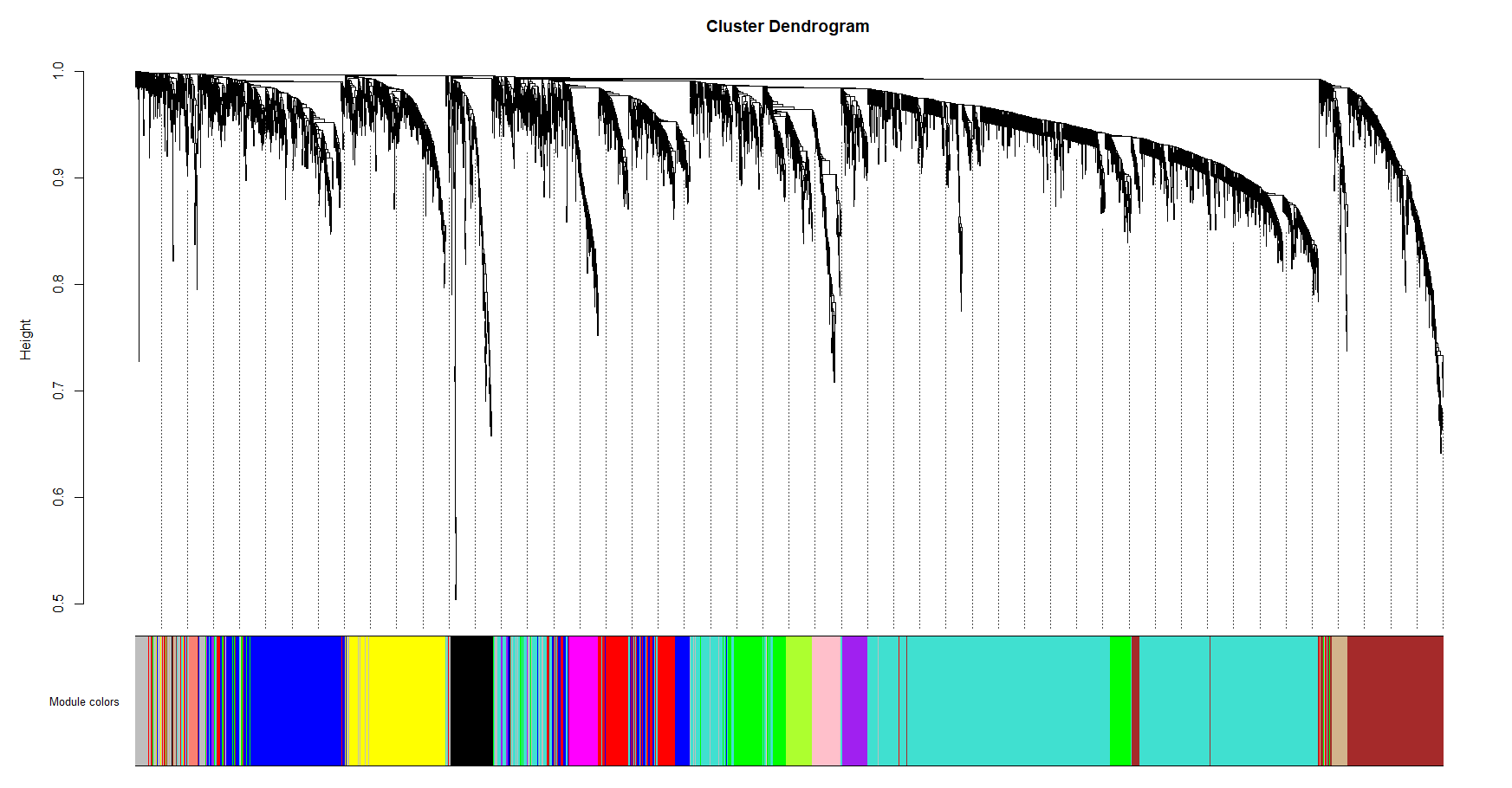
性状矩阵整理
这一步需要手动,把自己要观察的性状数值化,如果本来不是数字的指标,比如生存结局(生或死),要赋值,比如 0=活,1=死。这一步最后的矩阵整理为 datTraits,最后得到module和trait的相关性热图
MEs0 = moduleEigengenes(datExpr, moduleColors)$eigengenes # 计算MEMEs = orderMEs(MEs0) # 共表达基因order到一起moduleTraitCor = cor(MEs, datTraits, use = "p") # core!! 计算模块和性状相关性nSamples <- nrow(datExpr)moduleTraitPvalue = corPvalueStudent(moduleTraitCor, nSamples) # 计算模块和性状相关性p值textMatrix = paste(signif(moduleTraitCor, 2), "\n(",signif(moduleTraitPvalue, 1), ")", sep = "");dim(textMatrix) = dim(moduleTraitCor)textMatrix[1:3,1:3]# 画相关性热图par(mar = c(9, 10, 3, 3))labeledHeatmap(Matrix = moduleTraitCor,xLabels = colnames(datTraits),yLabels = colnames(MEs),ySymbols = colnames(MEs),colorLabels = FALSE,colors = blueWhiteRed(50),textMatrix = textMatrix,setStdMargins = FALSE,cex.text = 0.3,zlim = c(-1,1),main = paste("Module-trait relationships"))
提取感兴趣的模块
每个基因都会被赋予一个模块,通过下面这句可以提取基因的模块,并且赋予一个颜色
module = as.data.frame(net$colors)colnames(module)[1] = 'label'module$color = labels2colors(module$label)plotDendroAndColors(net$dendrograms[[1]], moduleColors[net$blockGenes[[1]]],"Module colors",dendroLabels = FALSE, hang = 0.03,addGuide = TRUE, guideHang = 0.05) #生成层次聚类树和模块对应图
模块PPI
这一部分比较需要配置

若有收获,就点个赞吧
0 人点赞
