take “ACC” for example
(一)
先建立Rproj,然后在工作目录下建立import文件夹以及import/ACC文件夹
从TCGA上需要下载四个文件:gdc-client.exe (TCGA官方下载工具)、表达矩阵、临床信息和注释文件
1.1 gdc-client
gdc-client.exe下载网址:https://gdc.cancer.gov/access-data/gdc-data-transfer-tool
gdc-client.exe下载好保存到Rproj的工作目录下(只是为了方便,理论上说,保存路径只要能知道在哪就行)
1.2 临床信息
临床信息&表达矩阵网址:https://portal.gdc.cancer.gov/repository
(看我红箭头指向的框起来的地方,下面的勾选信息就在这里选)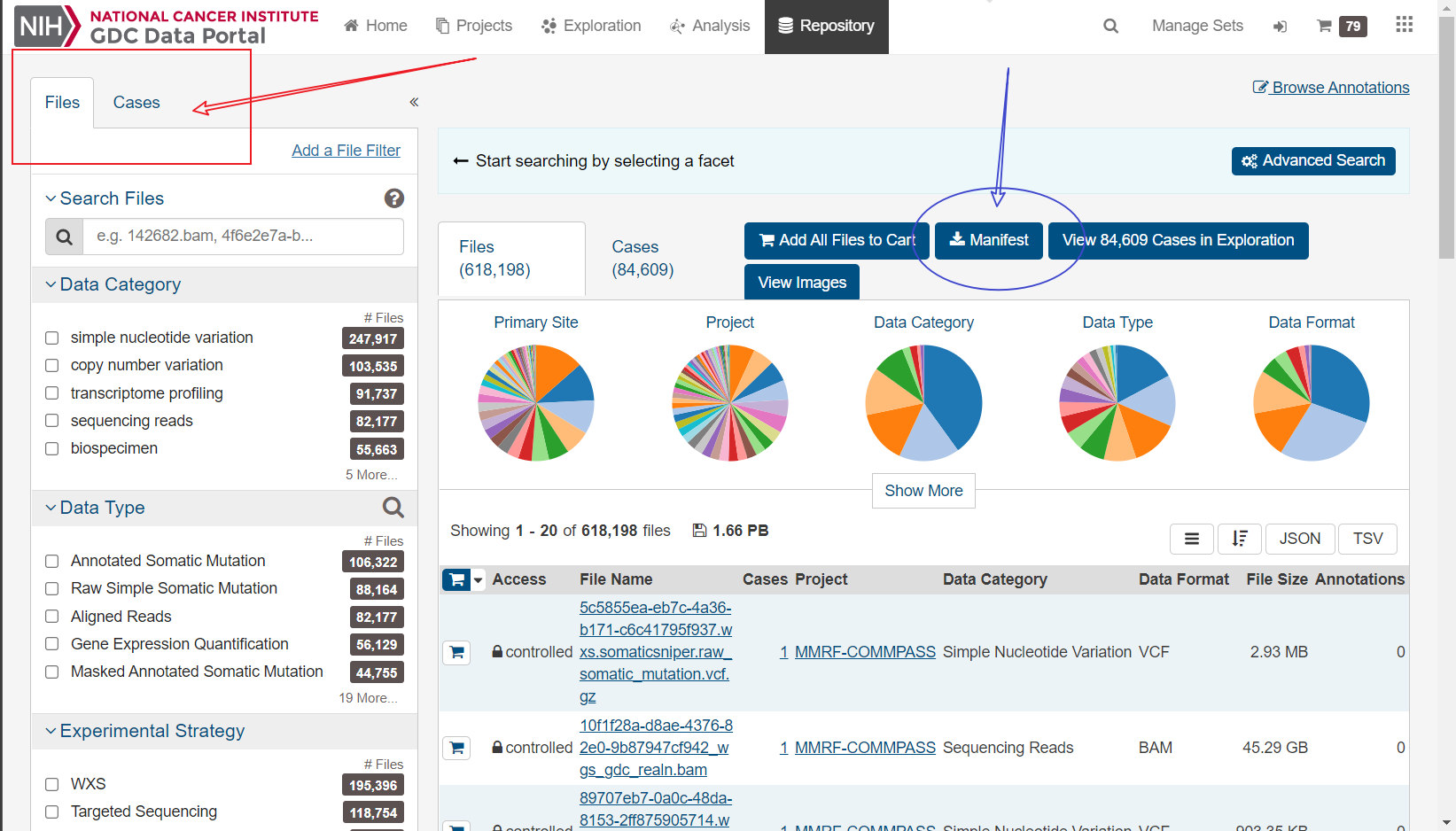
临床信息:需要勾选四个地方
Cases:Program勾TCGA; Project勾TCGA-ACC
Files:Data Category 勾 clinical;Data Format 勾 bcr xml
这四个勾完之后,点上图用蓝色箭头指向的Manifest,就可以得到临床信息的下载清单
(这时候只是下载了需要下载文件的清单,并没有下载文件)
文件名为 gdc_manifest_cl.txt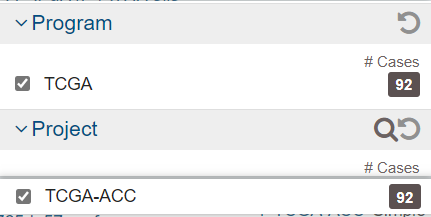

1.3 表达矩阵
表达矩阵:也需要勾选四个地方
Cases同上
Files:Data Category 勾 transcriptome profiling; Workflow Type 勾 HTSeq-Counts
这四个勾完之后,点上图用蓝色箭头指向的Manifest,就可以得到表达矩阵的下载清单
(这时候只是下载了需要下载文件的清单,并没有下载文件)
文件名为 gdc_manifest_exp.txt
1.4 注释文件
点击右侧的购物车,选“Add all files to the Cart”,然后右上角进入购物车,下载metadata.json文件
文件名是 metadata.json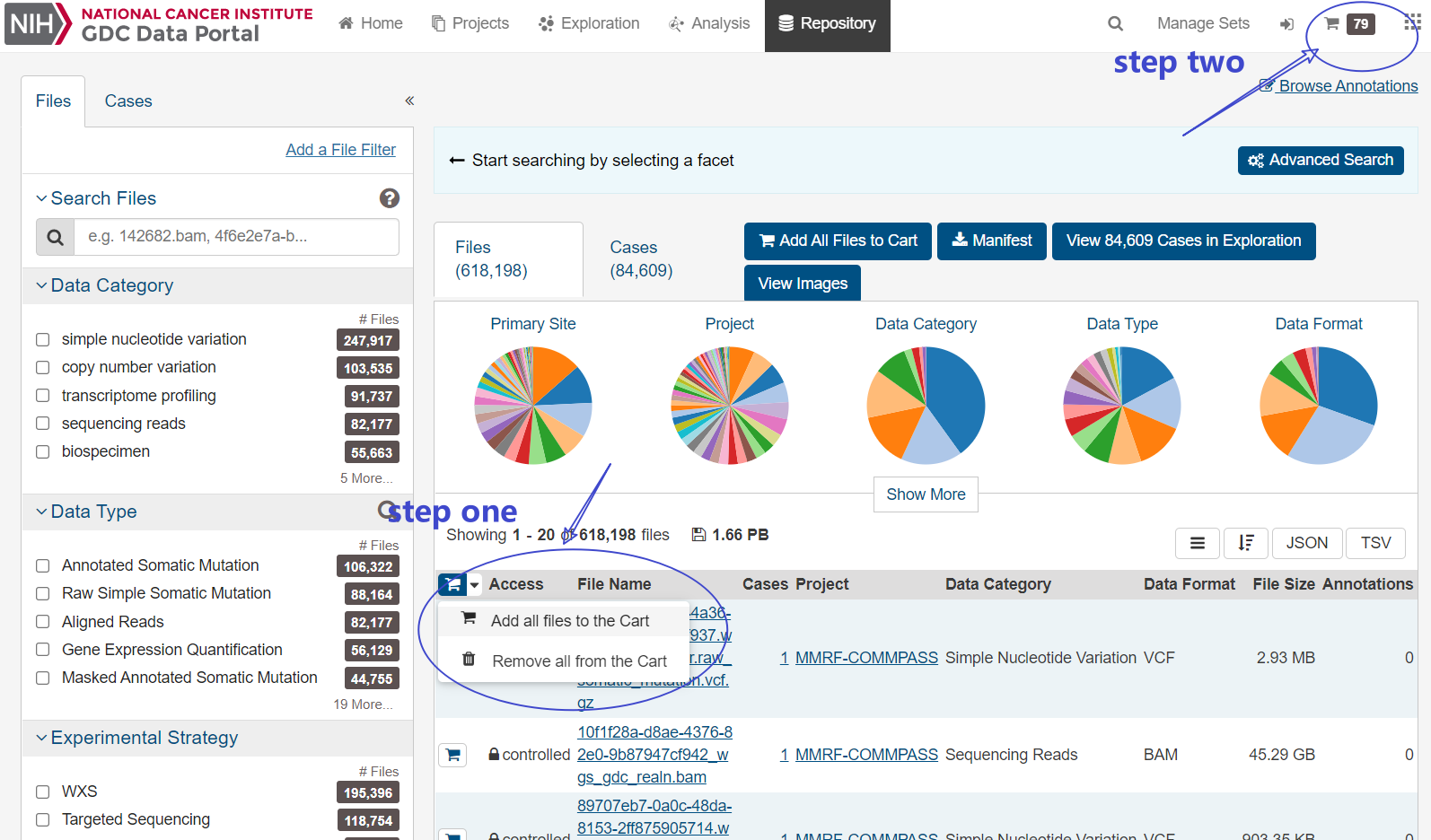
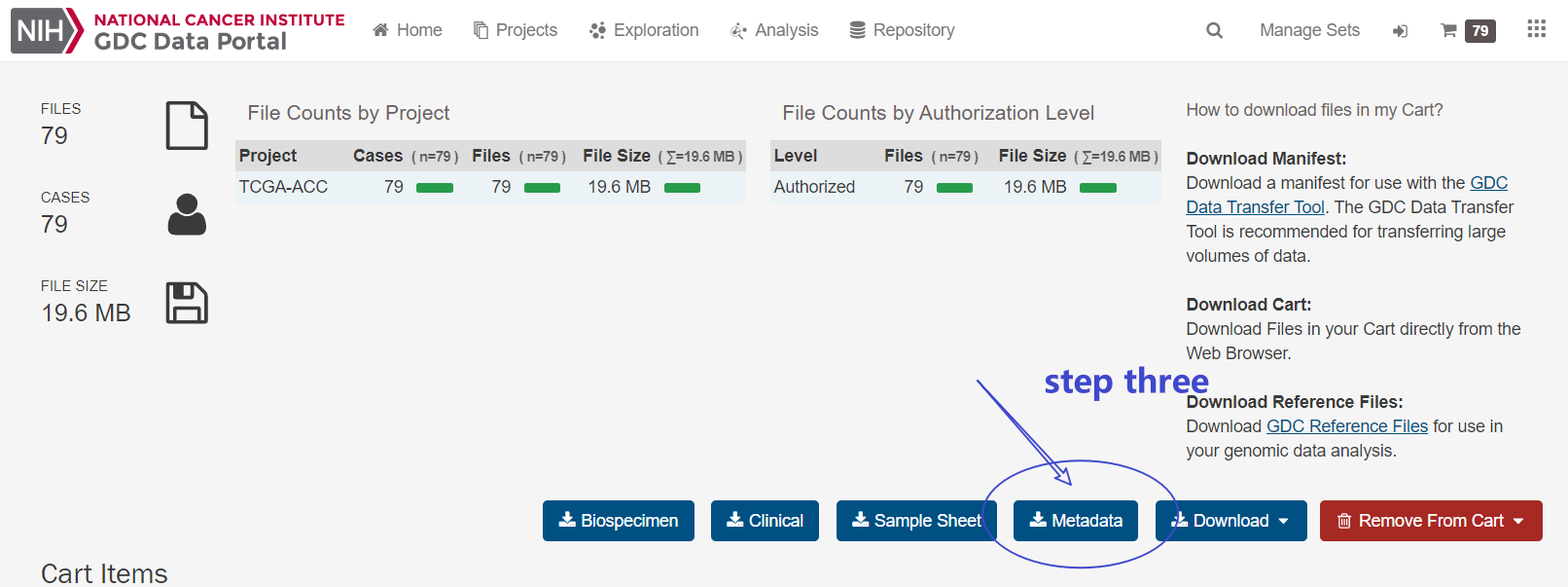
为了后面不改代码,都下在import/ACC里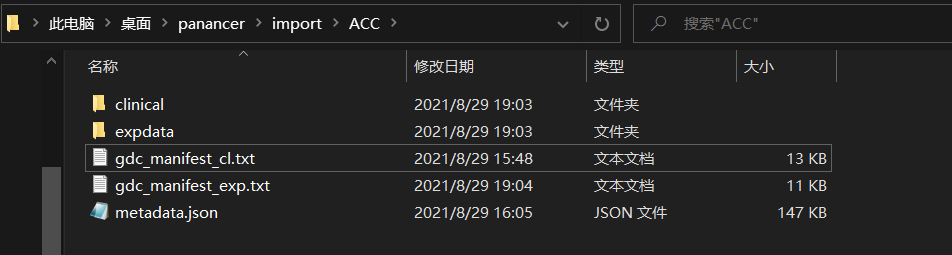
(二)
2.1 根据清单下载临床信息和表达矩阵
(由于我是电脑路径整理狂,所以在Rproj下建了很多文件,个人习惯)
下面的代码块除了 disease <- “ACC” ,其他全在建文件夹
理论上说,下面所有的代码只需要改一下 disease <- “ACC” 冒号里的癌症种类,其他的可以无脑运行(前提是上面的文件下载对了)
dir <- c("import", "outport", "Rscript", "Rdata", "figures")for (i in 1:length(dir)){if(!dir.exists(dir[i])) dir.create(dir[i])}disease <- "ACC" # 只改这里if(!dir.exists(paste0("import/", disease)))dir.create(paste0("import/", disease))if(!dir.exists(paste0("import/", disease, "/clinical")))dir.create(paste0("import/", disease, "/clinical"))if(!dir.exists(paste0("import/", disease, "/expdata")))dir.create(paste0("import/", disease, "/expdata"))
下两行代码在terminal完成 可能会报错 要么就是gdc-client.exe 需要换个新版本的,要么就找个网好的地方多下几次
如果做的是其他癌症,那么ACC的地方需要改!!!
下面这两行如果报错,就把/变成\,一个linux 一个windowns而已
./gdc-client.exe download -m ./import/ACC/gdc_manifest_cl.txt -d ./import/ACC/clinical/./gdc-client.exe download -m ./import/ACC/gdc_manifest_expdata.txt -d ./import/ACC/expdata/
terminal位置:红箭头指向的地方(善用tab补齐)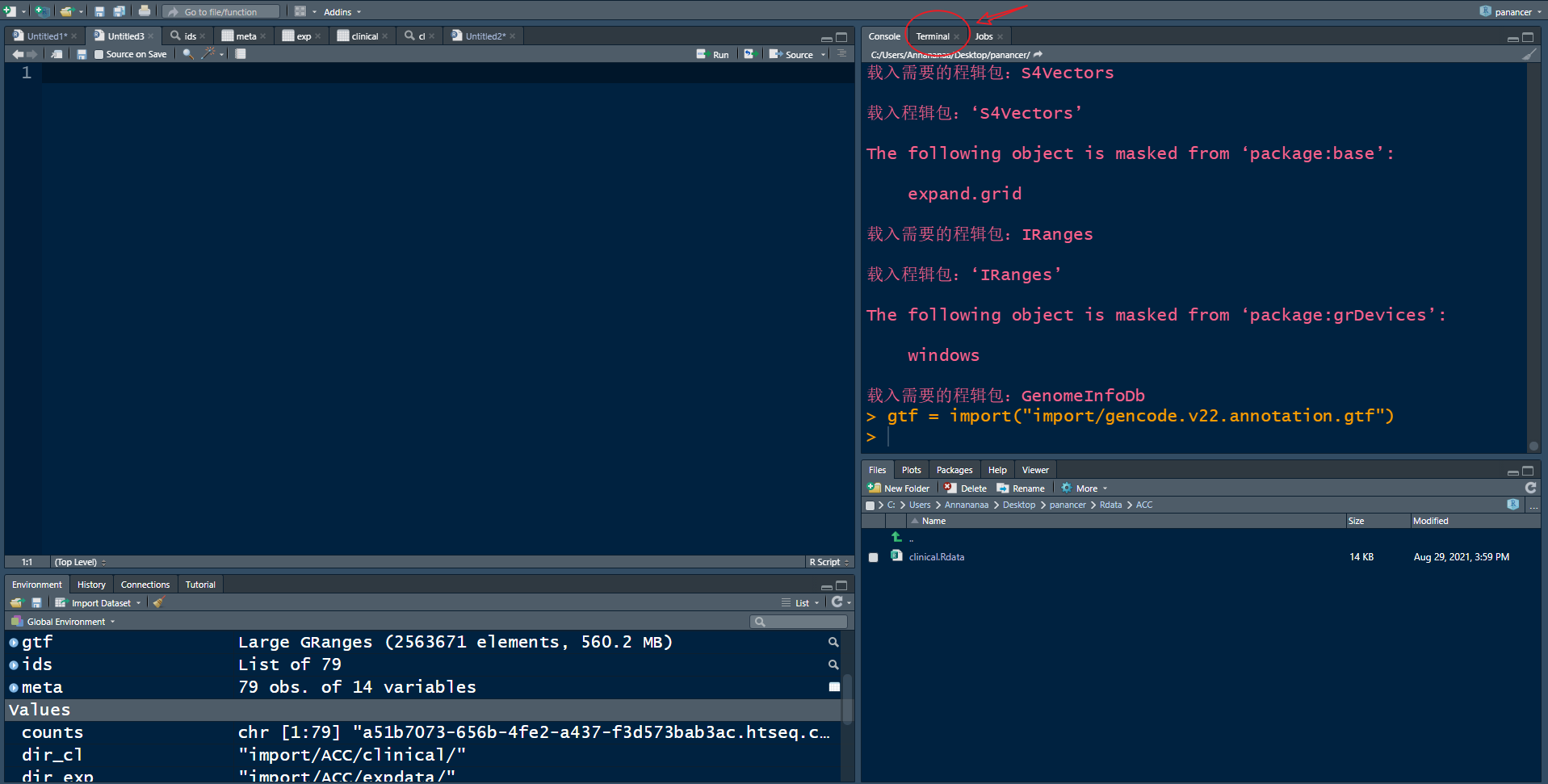
由于我的路径太长了,建个变量,顺便检查一下下载文件是否完整
dir_cl <- paste0("import/", disease, "/clinical/")dir_exp <- paste0("import/", disease, "/expdata/")length(dir(dir_cl))length(dir(dir_exp))
2.2 临床信息整理
用到了for循环,for循环其实还挺常用的
最后得到的clinical就是ACC所有病人的所有临床信息了
library(XML) #读取xml的r包xmls <- dir(dir_cl, pattern = "*.xml$", recursive = T) #匹配以xml结尾的clinical中的所有文件cl <- list()for(i in 1:length(xmls)){result <- xmlParse(paste0(dir_cl,xmls[[i]])) #补充读取xml的路径rootnode <- xmlRoot(result) #查看结点数目 可用print(rootnode[1])查看每个节点的信息cl[[i]] <- xmlToDataFrame(rootnode[2]) #将xml读取的东西变成数据框}clinical <- do.call(rbind, cl) # 将所有clinical 的list集合为一个数据框dim(clinical)if(!dir.exists(paste0("Rdata/", disease)))dir.create(paste0("Rdata/", disease))save(clinical, file = paste0("Rdata/", disease, "/clinical.Rdata"))
2.3 表达矩阵整理
counts <- dir(dir_exp, pattern = "*.htseq.counts.gz$", recursive = T) #匹配以xml结尾的clinical中的所有文件expdata <- list()for(i in 1:length(counts)){expdata[[i]] <- read.table(paste0(dir_exp,counts[[i]]),row.names = 1,sep = "\t")}expdata <- do.call(cbind,expdata)dim(expdata)
表达矩阵每一行是一个基因,每一列是一位病人
这时候点开expdata会发现,没有列名,就需要下载的第四个文件,注释信息上场
下面有个match函数,特别常用;stringr这个包也有很多好用的函数
meta <- jsonlite::fromJSON(paste0("import/", disease, "/metadata.json"))colnames(meta)ids <- meta$associated_entitiesID <- sapply(ids,function(x){x[,1]})file2id <- data.frame(file_name = meta$file_name,ID = ID)counts <- stringr::str_split(counts,"/",simplify = T)[,2]table(counts %in% file2id$file_name)table(counts == file2id$file_name) # 两者数目对应 但是顺序不一致file2id <- file2id[match(counts, file2id$file_name),]colnames(expdata) <- file2id$ID
这时候又发现 expdata的行名是Ensembl ID,先转换成 gene symbol
TCGA 有自己的转换方式,下载一个gtf文件
网址:https://gdc.cancer.gov/about-data/gdc-data-processing/gdc-reference-files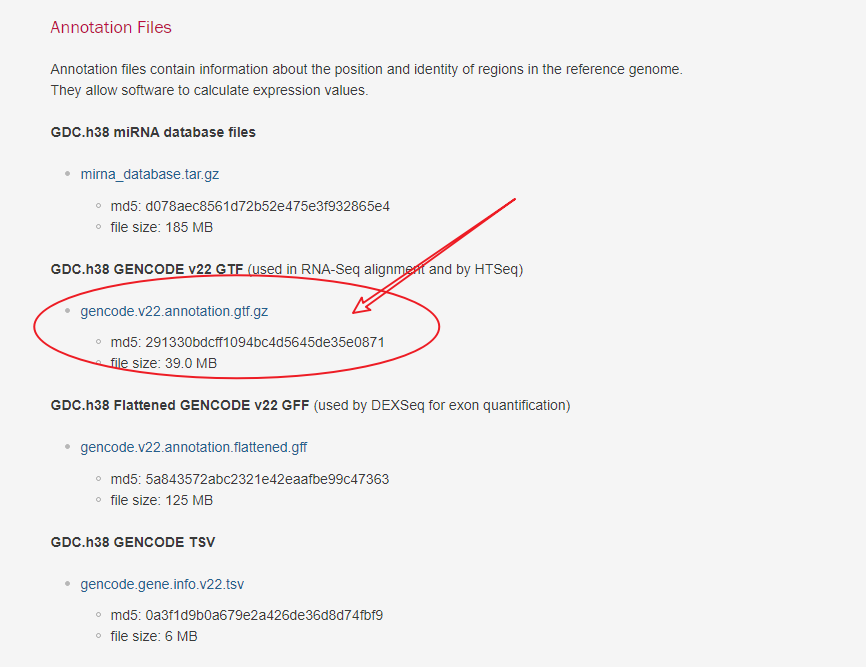
然后无脑运行下面的id转换(懒得解释了,有R语言基础应该可以看懂)
library(rtracklayer)gtf <- import("import/gencode.v22.annotation.gtf")class(gtf)gtf <- as.data.frame(gtf)colnames(gtf)table(gtf$type)#step2:先筛选出gene对应的行gtf_gene <- gtf[gtf$type=="gene",]an <- gtf_gene[,c("gene_name","gene_id","gene_type")]expdata <- expdata[rownames(expdata) %in% an$gene_id,]an <- an[match(rownames(expdata),an$gene_id),]identical(an$gene_id,rownames(expdata))k <- !duplicated(an$gene_name);table(k)an <- an[k,]expdata <- expdata[k,]rownames(expdata) <- an$gene_name
病人分组信息整理,TCGA 病人ID的命名规则:14~15位是分组信息,小于10是tumor,大于等于10是normal
不太巧,ACC的79位全是癌症病人(我看这个数据集少才用它的,妹想到…)
table(stringr::str_sub(colnames(expdata),14 ,15))Group <- ifelse(stringr::str_sub(colnames(expdata),14 ,15)<10, "tumor", "normal")
到这里expdata大概就整理完了,有一个问题就是,在expdata里有很多表达量几乎为0的基因,这些基因本身没什么意义,但是由于表达量过低,有可能在后续分析的时候,结果反而显著,因此最好筛选一下
筛选的标准因人而异吧,我是用counts数在50%的病人里>10筛选的,如果觉得太严格,可以放宽一些
number <- 0.5*ncol(expdata)expdata <- expdata[apply(expdata, 1, function(x) sum(x > 10) >number), ]save(expdata, Group, file = paste0("Rdata/", disease, "/expdata.Rdata"))
初步的下载工作就完成了~
整理好累…
(三)
代码打包
one more time: 注意官网下载文件放的位置!
dir <- c("import", "outport", "Rscript", "Rdata", "figures")for (i in 1:length(dir)){if(!dir.exists(dir[i])) dir.create(dir[i])}get_disease_info = function(diseasename){disease <- diseasenameif(!dir.exists(paste0("import/", disease)))dir.create(paste0("import/", disease))if(!dir.exists(paste0("import/", disease, "/clinical")))dir.create(paste0("import/", disease, "/clinical"))if(!dir.exists(paste0("import/", disease, "/expdata")))dir.create(paste0("import/", disease, "/expdata"))# 下载临床信息和表达矩阵system(paste0("./gdc-client.exe download -m ./import/",disease,"/gdc_manifest_cl.txt -d ./import/",disease,"/clinical/"))system(paste0("./gdc-client.exe download -m ./import/",disease,"/gdc_manifest_exp.txt -d ./import/",disease,"/expdata/"))dir_cl <- paste0("import/", disease, "/clinical/")dir_exp <- paste0("import/", disease, "/expdata/")## 临床信息整理library(XML)xmls <- dir(dir_cl, pattern = "*.xml$", recursive = T)cl <- list()for(i in 1:length(xmls)){result <- xmlParse(paste0(dir_cl,xmls[[i]]))rootnode <- xmlRoot(result)cl[[i]] <- xmlToDataFrame(rootnode[2])}clinical <- do.call(rbind, cl)if(!dir.exists(paste0("Rdata/", disease)))dir.create(paste0("Rdata/", disease))save(clinical, file = paste0("Rdata/", disease, "/clinical.Rdata"))## 表达矩阵整理counts <- dir(dir_exp, pattern = "*.htseq.counts.gz$", recursive = T)expdata <- list()for(i in 1:length(counts)){expdata[[i]] <- read.table(paste0(dir_exp,counts[[i]]),row.names = 1,sep = "\t")}expdata <- do.call(cbind,expdata)# 添加列名meta <- jsonlite::fromJSON(paste0("import/", disease, "/metadata.json"))ids <- meta$associated_entitiesID <- sapply(ids,function(x){x[,1]})file2id <- data.frame(file_name = meta$file_name,ID = ID)counts <- stringr::str_split(counts,"/",simplify = T)[,2]file2id <- file2id[match(counts, file2id$file_name),]colnames(expdata) <- file2id$ID# 将行名换成gene symbol# TCGA有自己的id转换方法library(rtracklayer)gtf <- import("import/gencode.v22.annotation.gtf")gtf <- as.data.frame(gtf)gtf_gene <- gtf[gtf$type=="gene",]an <- gtf_gene[,c("gene_name","gene_id","gene_type")]expdata <- expdata[rownames(expdata) %in% an$gene_id,]an <- an[match(rownames(expdata),an$gene_id),]k <- !duplicated(an$gene_name);table(k)an <- an[k,]expdata <- expdata[k,]rownames(expdata) <- an$gene_nameGroup <- ifelse(stringr::str_sub(colnames(expdata),14 ,15)<10, "tumor", "normal")# 过滤低表达基因(玄学)number <- 0.5*ncol(expdata)expdata <- expdata[apply(expdata, 1, function(x) sum(x > 10) >number), ]save(expdata, Group, file = paste0("Rdata/", disease, "/expdata.Rdata"))}get_disease_info('ACC')

若有收获,就点个赞吧
0 人点赞
