用 R 提取代表性转录本
前言:
上上篇文章讲了从 GTF 文件的提取代表性的转录本 gtf 信息:提取代表转录本之 gencode,如果想获得代表性转录本序列的话,可以使用 gffread 软件取获得 cDNA 序列。
也可以直接下载转录组序列,但是里面包含了同一个基因的多个转录本,我们可以筛选出代表性的转录本。筛选原则与上面的文章有一些不同,我们先看下面这张图:
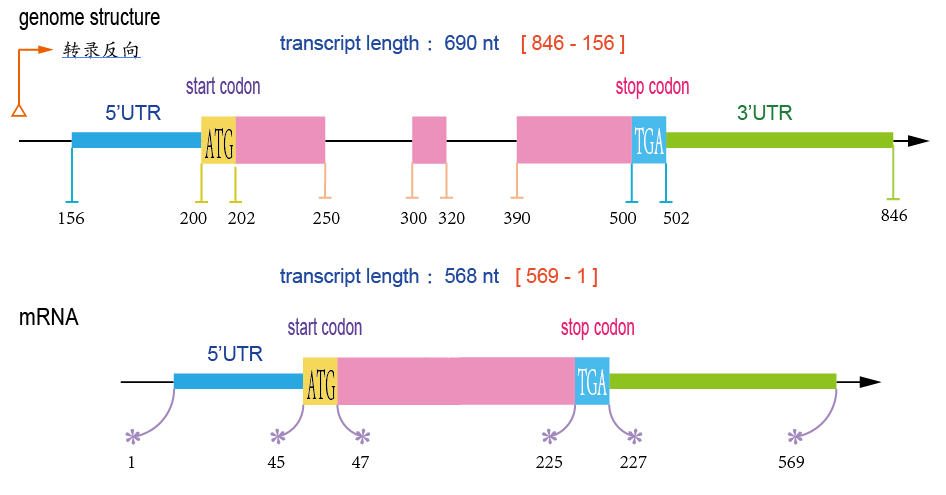
基于 gtf 注释文件挑选的最长转录本的长度是包含了内含子的长度,这样选择不能真实反应转录本的实际长度,在选择 CDS 最长的前提下,再选择转录本外显子的总长度最长更加合理。
提取实操:
1、数据准备
首先去 gencode 数据库下载蛋白编码基因的转录组序列文件:
# 下载$ wget http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_38/gencode.v38.pc_transcripts.fa.gz# 查看有多少个转录本序列$ zless gencode.v38.pc_transcripts.fa.gz | grep '>' | wc -l106143# 简单查看前面几行$ zless gencode.v38.pc_transcripts.fa.gz | head>ENST00000641515.2|ENSG00000186092.7|OTTHUMG00000001094.4|OTTHUMT00000003223.4|OR4F5-201|OR4F5|2618|UTR5:1-60|CDS:61-1041|UTR3:1042-2618|CCCAGATCTCTTCAGTTTTTATGCCTCATTCTGTGAAAATTGCTGTAGTCTCTTCCAGTTATGAAGAAGGTAACTGCAGAGGCTATTTCCTGGAATGAATCAACGAGTGAAACGAATAACTCTATGGTGACTGAATTCATTTTTCTGGGTCTCTCTGATTCTCAGGAACTCCAGACCTTCCTATTTATGTTGTTTTTTGTATTCTATGGAGGAATCGTGTTTGGAAACCTTCTTATTGTCATAACAGTGGTATCTGACTCCCACCTTCACTCTCCCATGTACTTCCTGCTAGCCAACCTCTCACTCATTGATCTGTCTCTGTCTTCAGTCACAGCCCCCAAGATGATTACTGACTTTTTCAGCCAGCGCAAAGTCATCTCTTTCAAGGGCTGCCTTGTTCAGATATTTCTCCTTCACTTCTTTGGTGGGAGTGAGATGGTGATCCTCATAGCCATGGGCTTTGACAGATATATAGCAATATGCAAGCCCCTACACTACACTACAATTATGTGTGGCAACGCATGTGTCGGCATTATGGCT
ENST00000641515.2|ENSG00000186092.7|OTTHUMG00000001094.4|OTTHUMT00000003223.4|OR4F5-201|OR4F5|2618|UTR5:1-60|CDS:61-1041|UTR3:1042-2618|
每个序列的 ID 由这么一长串内容组成,分别是:
- ENST00000641515.2 : ensembl 的转录本 id
- ENSG00000186092.7 : ensembl 的基因 id
- OTTHUMG00000001094.4 : havana 的基因 id
- OTTHUMT00000003223.4 : havana 的转录本 id
- OR4F5-201 : 转录本名称
- OR4F5 : 基因名
- 2618 : 转录本序列总长度
- UTR5:1-60 : 转录本 5’UTR 的位置
- CDS:61-1041 : 转录本 CDS 的位置
- UTR3:1042-2618 : 转录本 3’UTR 的位置
我还想重新命名每个转录本序列的 ID,只保留基因名、gene_id、transcript_id、transcript_name、CDS 起始位置和终止位置、转录本长度,类似于下面这种更加简洁一点:
>OR4F5_ENSG00000186092.7_ENST00000641515.2_OR4F5-201_61_1041_2618>OR4F29_ENSG00000284733.2_ENST00000426406.4_OR4F29-201_1_939_939>OR4F16_ENSG00000284662.1_ENST00000332831.4_OR4F16-201_20_958_995
把转录组序列名字输出保存到一个文件里,同时去除 havana 的基因 id 和转录本 id:
# 按 | 字符分割$ zless gencode.v38.pc_transcripts.fa.gz |grep '>' |sed 's/|/\t/g' |cut -f 1,2,5,6,7,8,9,10 > name.txt# 查看内容$ head -3 name.txt>ENST00000641515.2 ENSG00000186092.7 OR4F5-201 OR4F5 2618 UTR5:1-60 CDS:61-1041 UTR3:1042-2618>ENST00000426406.4 ENSG00000284733.2 OR4F29-201 OR4F29 939 CDS:1-939>ENST00000332831.4 ENSG00000284662.1 OR4F16-201 OR4F16 995 UTR5:1-19 CDS:20-958 UTR3:959-995
2、改名操作
导入 R 里:
# 设置工作路径setwd('C:/Users/admin/Desktop/test/')# 读取id数据all_name <- read.delim('name.txt',header = F)# 添加列名colnames(all_name) <- c('tt_id','gene_id','tt_name','gene_name','tt_length','info1','info2','info3')# 查看行数nrow(all_name)[1] 106143
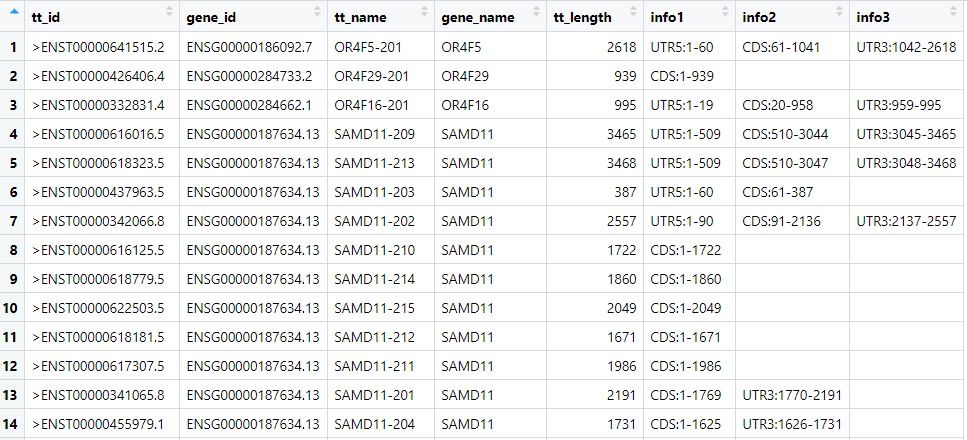
可以看到最后 3 列相同的内容不在一列里,这是因为有些转录本只有 CDS 区或者 5’UTR 和 CDS 区,我们需要增加一列都是 CDS 区的位置信息:
# 添加cds列library(stringr)# 新建储存cds信息的listcds_list <- list()# 循环for (i in 1:nrow(all_name)) {# 提取数据all_name_filter <- all_name[i,]# 分割info1info_1 <- unlist(strsplit(all_name_filter$info1,split = ':|-'))if (info_1[1] == 'UTR5') {# 如果第一个是UTR5,则取第二个元素cds_list[[i]] <- all_name_filter$info2}else if(info_1[1] == 'CDS') {# 如果第一个是CDS,则取第一个元素cds_list[[i]] <- all_name_filter$info1}else {}}# 查看个数length(cds_list)[1] 106143# 合并cds数据cds_res <- do.call('rbind',cds_list)# 储存在新数据里all_name_cds <- all_nameall_name_cds$cds <- cds_res# 查看内容head(all_name_cds,4)tt_id gene_id tt_name gene_name tt_length info1 info2 info31 >ENST00000641515.2 ENSG00000186092.7 OR4F5-201 OR4F5 2618 UTR5:1-60 CDS:61-1041 UTR3:1042-26182 >ENST00000426406.4 ENSG00000284733.2 OR4F29-201 OR4F29 939 CDS:1-9393 >ENST00000332831.4 ENSG00000284662.1 OR4F16-201 OR4F16 995 UTR5:1-19 CDS:20-958 UTR3:959-9954 >ENST00000616016.5 ENSG00000187634.13 SAMD11-209 SAMD11 3465 UTR5:1-509 CDS:510-3044 UTR3:3045-3465cds1 CDS:61-10412 CDS:1-9393 CDS:20-9584 CDS:510-3044#################################### 改名# 新建储存添加列名的listst_sp_list <- list()# 循环for (i in 1:nrow(all_name_cds)) {# 提取数据all_name_cds_filter <- all_name_cds[i,]# 分割info1info_cds <- unlist(strsplit(all_name_cds_filter$cds,split = ':|-'))# 去掉tt_id的 > 符号ttid <- unlist(strsplit(all_name_cds_filter$tt_id,split = '>'))[2]# 添加改名列all_name_cds_filter$new_name <- paste(all_name_cds_filter$gene_name,all_name_cds_filter$gene_id,ttid,all_name_cds_filter$tt_name,info_cds[2],info_cds[3],all_name_cds_filter$tt_length,sep = '_')# 储存结果st_sp_list[[i]] <- all_name_cds_filter}length(st_sp_list)[1] 106143# 合并数据st_sp_res <- do.call('rbind',st_sp_list)# 查看内容head(st_sp_res$new_name,4)[1] "OR4F5_ENSG00000186092.7_ENST00000641515.2_OR4F5-201_61_1041_2618"[2] "OR4F29_ENSG00000284733.2_ENST00000426406.4_OR4F29-201_1_939_939"[3] "OR4F16_ENSG00000284662.1_ENST00000332831.4_OR4F16-201_20_958_995"[4] "SAMD11_ENSG00000187634.13_ENST00000616016.5_SAMD11-209_510_3044_3465"
这样我们就成功的改好了名字!接下来我们需要读入转录组序列,把它们重新命名。
# 读取转录组序列fasta文件# 安装读取fasta文件的R包BiocManager::install('seqinr')library(seqinr)fasta <- read.fasta('gencode.v38.pc_transcripts.fa.gz',seqtype = 'DNA',forceDNAtolower = F)# fasta数据类型class(fasta)[1] "list"# 查看命名前的名称names(fasta)[1][1] "ENST00000641515.2|ENSG00000186092.7|OTTHUMG00000001094.4|OTTHUMT00000003223.4|OR4F5-201|OR4F5|2618|UTR5:1-60|CDS:61-1041|UTR3:1042-2618|"# 命名names(fasta) <- st_sp_res$new_name# 查看命名后的名称names(fasta)[1][1] "OR4F5_ENSG00000186092.7_ENST00000641515.2_OR4F5-201_61_1041_2618"# 保存命名后的数据write.fasta(sequences = fasta,names = names(fasta),file.out = 'name_changed.fa')
3、提取代表性转录本
主要思路,我们已经获得了转录本长度的信息,只要计算每个转录本的 CDS 区长度即可,然后按 CDS 列和转录本长度列降序排序取第一个就行了。
library(tidyverse)# 提取代表性转录本# 新建储存筛选出代表性转录本的信息repsentive_tspt <- list()# genegene <- unique(st_sp_res$gene_name)# 循环for (g in gene) {# 筛选geneginfo <- st_sp_res %>% filter(gene_name == g)# 添加cds length# 切分cds列cds_list <- strsplit(ginfo$cds,split = ':|-')# 获取cds start和stop位置start <- sapply(cds_list, '[',2)stop <- sapply(cds_list, '[',3)# 计算赋值cds lengthginfo$cds_length <- as.numeric(stop) - as.numeric(start)# 先按cds_length列降序,再按tt_length列降序ginfo_order <- ginfo %>% arrange(desc(cds_length,tt_length))# 筛选transcripttid <- ginfo$tt_id[1]list_name <- paste(g,tid,sep = '_')# 取第一行元素保存repsentive_tspt[[list_name]] <- ginfo_order[1,]}# 查看筛选结果数量length(repsentive_tspt)[1] 20336repsentive_results <- do.call('rbind',repsentive_tspt)# 查看有多少个基因length(unique(repsentive_results$gene_id))[1] 20336# 查看有多少个转录本length(unique(repsentive_results$tt_id))[1] 20336# 查看结果head(repsentive_results,4)tt_id gene_id tt_name gene_name tt_length info1OR4F5_>ENST00000641515.2 >ENST00000641515.2 ENSG00000186092.7 OR4F5-201 OR4F5 2618 UTR5:1-60OR4F29_>ENST00000426406.4 >ENST00000426406.4 ENSG00000284733.2 OR4F29-201 OR4F29 939 CDS:1-939OR4F16_>ENST00000332831.4 >ENST00000332831.4 ENSG00000284662.1 OR4F16-201 OR4F16 995 UTR5:1-19SAMD11_>ENST00000616016.5 >ENST00000618323.5 ENSG00000187634.13 SAMD11-213 SAMD11 3468 UTR5:1-509info2 info3 cdsOR4F5_>ENST00000641515.2 CDS:61-1041 UTR3:1042-2618 CDS:61-1041OR4F29_>ENST00000426406.4 CDS:1-939OR4F16_>ENST00000332831.4 CDS:20-958 UTR3:959-995 CDS:20-958SAMD11_>ENST00000616016.5 CDS:510-3047 UTR3:3048-3468 CDS:510-3047new_name cds_lengthOR4F5_>ENST00000641515.2 OR4F5_ENSG00000186092.7_ENST00000641515.2_OR4F5-201_61_1041_2618 980OR4F29_>ENST00000426406.4 OR4F29_ENSG00000284733.2_ENST00000426406.4_OR4F29-201_1_939_939 938OR4F16_>ENST00000332831.4 OR4F16_ENSG00000284662.1_ENST00000332831.4_OR4F16-201_20_958_995 938SAMD11_>ENST00000616016.5 SAMD11_ENSG00000187634.13_ENST00000618323.5_SAMD11-213_510_3047_3468 2537
接下来该怎么从筛选的结果中取提取 fasta 数据呢?
# 读取改名后的fastanamed_fasta <- read.fasta('name_changed.fa',seqtype = 'DNA',forceDNAtolower = F)# 对代表性转录本循环tar_name <- repsentive_results$new_name# 建立储存筛选结果的listtarget_falist <- list()# 循环
for (i in tar_name) {choose <- i %in% names(named_fasta)if ("TRUE" %in% choose) {target_falist[i] <- named_fasta[i]}else{}}# 查看筛选结果数量length(target_falist)[1] 20336# 保存筛选的数据write.fasta(sequences = target_falist,names = names(target_falist),file.out = 'test_transcript.fa')
查看数据内容:
$ head test_transcript.fa>OR4F5_ENSG00000186092.7_ENST00000641515.2_OR4F5-201_61_1041_2618CCCAGATCTCTTCAGTTTTTATGCCTCATTCTGTGAAAATTGCTGTAGTCTCTTCCAGTTATGAAGAAGGTAACTGCAGAGGCTATTTCCTGGAATGAATCAACGAGTGAAACGAATAACTCTATGGTGACTGAATTCATTTTTCTGGGTCTCTCTGATTCTCAGGAACTCCAGACCTTCCTATTTATGTTGTTTTTTGTATTCTATGGAGGAATCGTGTTTGGAAACCTTCTTATTGTCATAACAGTGGTATCTGACTCCCACCTTCACTCTCCCATGTACTTCCTGCTAGCCAACCTCTCACTCATTGATCTGTCTCTGTCTTCAGTCACAGCCCCCAAGATGATTACTGACTTTTTCAGCCAGCGCAAAGTCATCTCTTTCAAGGGCTGCCTTGTTCAGATATTTCTCCTTCACTTCTTTGGTGGGAGTGAGATGGTGATCCTCATAGCCATGGGCTTTGACAGATATATAGCAATATGCAAGCCCCTACACTACACTACAATTATGTGTGGCAACGCATGTGTCGGCATTATGGCT
OK!搞定!

若有收获,就点个赞吧
0 人点赞
