提取代表转录本之 gencode
Introduction
一个基因存在一个或多个转录本(variants),后续我们想研究某个基因的话,那么如何选取哪个转录本进行研究?比如引物设计等,或者绘图。为了减少工作量可以选取基因的代表性的转录本(Representative transcript)进行研究,那么如何选取合适或合理的 代表转录本 呢?
- 1、选转录本最长的
- 2、选表达量最高的
- 3、选外显子数量最多的
貌似第一种选取最长的转录本作为研究是最多的,很多文献里也使用过,至于合不合理都有一定的缺点,也许发挥主要功能的不一定是最长的那个转录本,也可能不同转录本发挥着不一样的功能,但应该都是少数情况,大部分最长的转录本发挥主要功能。
成熟的 mRNA 要想发挥功能主要依赖于 CDS 区翻译出来的蛋白,成熟的蛋白是一切功能的承载者,基于此,选取代表转录本可以基于两个选择:
a、选取 CDS 区最长的 b、其次再选取最长的转录本
我们从 gencode 数据库下载 human 的注释文件,从里面提取 Representative transcript 再导出筛选出代表转录本的 gtf 文件。
开始分析
下载注释文件:
# wget 下载$ wget http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_38/gencode.v38.annotation.gtf.gz# 解压$ gunzip gencode.v38.annotation.gtf.gz# 查看一下内容$ less -S gencode.v38.annotation_human.gtf| head -10##description: evidence-based annotation of the human genome (GRCh38), version 38 (Ensembl 104)##provider: GENCODE##contact: gencode-help@ebi.ac.uk##format: gtf##date: 2021-03-12chr1 HAVANA gene 11869 14409 . + . gene_id "ENSG00000223972.5"; gene_type "transcribed_unprocessed_pseudogene"; gene_name "DDX11L1"; level 2; hgnc_id "HGNC:37102"; havana_gene "OTTHUMG00000000961.2";chr1 HAVANA transcript 11869 14409 . + . gene_id "ENSG00000223972.5"; transcript_id "ENST00000456328.2"; gene_type "transcribed_unprocessed_pseudogene"; gene_name "DDX11L1"; transcript_type "processed_transcript"; transcript_name "DDX11L1-202"; level 2; transcript_support_level "1"; hgnc_id "HGNC:37102"; tag "basic"; havana_gene "OTTHUMG00000000961.2"; havana_transcript "OTTHUMT00000362751.1";chr1 HAVANA exon 11869 12227 . + . gene_id "ENSG00000223972.5"; transcript_id "ENST00000456328.2"; gene_type "transcribed_unprocessed_pseudogene"; gene_name "DDX11L1"; transcript_type "processed_transcript"; transcript_name "DDX11L1-202"; exon_number 1; exon_id "ENSE00002234944.1"; level 2; transcript_support_level "1"; hgnc_id "HGNC:37102"; tag "basic"; havana_gene "OTTHUMG00000000961.2"; havana_transcript "OTTHUMT00000362751.1";chr1 HAVANA exon 12613 12721 . + . gene_id "ENSG00000223972.5"; transcript_id "ENST00000456328.2"; gene_type "transcribed_unprocessed_pseudogene"; gene_name "DDX11L1"; transcript_type "processed_transcript"; transcript_name "DDX11L1-202"; exon_number 2; exon_id "ENSE00003582793.1"; level 2; transcript_support_level "1"; hgnc_id "HGNC:37102"; tag "basic"; havana_gene "OTTHUMG00000000961.2"; havana_transcript "OTTHUMT00000362751.1";chr1 HAVANA exon 13221 14409 . + . gene_id "ENSG00000223972.5"; transcript_id "ENST00000456328.2"; gene_type "transcribed_unprocessed_pseudogene"; gene_name "DDX11L1"; transcript_type "processed_transcript"; transcript_name "DDX11L1-202"; exon_number 3; exon_id "ENSE00002312635.1"; level 2; transcript_support_level "1"; hgnc_id "HGNC:37102"; tag "basic"; havana_gene "OTTHUMG00000000961.2"; havana_transcript "OTTHUMT00000362751.1";
导入 gtf 文件
# 安装加载R包,读取gtf文件BiocManager::install('rtracklayer')library(tidyverse)library(rtracklayer)# 设置工作路径setwd('C:\\Users\\admin\\Desktop\\test\\')# 读取gtf文件gtf <- import('gencode.v38.annotation_human.gtf')# 转为data.framemy_gtf <- data.frame(gtf)
筛选
我们选取蛋白编码基因(protein coding)和 CCDS 共识基因(member of the consensus CDS gene set, confirming coding regions between ENSEMBL, UCSC, NCBI and HAVANA)。
# 筛选pt_gtf <- my_gtf %>% filter(gene_type == 'protein_coding',tag == 'CCDS')# 检查基因数量gene_name <- unique(pt_gtf$gene_name)length(gene_name)[1] 18808# 筛选出18808个基因
分析思路

因为我们有 18808 个基因需要循环走一遍,需要较长时间,所以在测试是可以取小样本测试,比如取 100 或 200 个基因测试即可。
提取代表性转录本
下面是循环体代码:
# 建一个储存结果信息的listlongest_gtf <- list()# 筛选# 测试100个基因gene_na <- gene_name[1:100]for (g in gene_na){# 挑选基因ginfo <- pt_gtf %>% filter(gene_name == g)# 提取转录本tinfo <- ginfo %>% filter(type == 'transcript')# 提取转录本数量ntspt <- nrow(tinfo)# 如果只有一个转录本if(ntspt == 1){# 提取只有一个转录本的转录本名one_tname <- tinfo$transcript_name# 提取基因和转录本信息gene_line <- my_gtf %>% filter(gene_name == g,type == 'gene')one_tspt <- my_gtf %>% filter(gene_name == g,transcript_name == one_tname)# 合并gene和转录本信息last_res <- rbind(gene_line,one_tspt)# 保存结果到list中longest_gtf[[g]] <- last_res}else{# 如果有多个转录本# 提取转录本名字tspt_name <- tinfo$transcript_name# 循环每个转录本# 储存转录本cds长度的listcds_length_all <- list()# 循环for(tname in tspt_name){mtinfo <- ginfo %>% filter(transcript_name == tname,type == 'CDS')# 计算cds区总长度cds_len <- sum(mtinfo$width)# 储存在list中cds_length_all[[tname]] <- cds_len}# 转化类型为向量a <- unlist(cds_length_all)# 查看是否有cds长度相等的转录本equal_cds <- duplicated(a)# 循环if("TRUE" %in% equal_cds){# 如果有相同cds长度相等的转录本就取转录本最长的# 获取最大cds长度max_num <- max(a)# 提取最大最大cds长度对应的转录本名称tn <- data.frame(a)tspt_name <- rownames(tn %>% filter(a == max_num))# 提取tinfo信息tspt_longest_info <- tinfo[which(tinfo$transcript_name %in% tspt_name),]# 按转录本长度降序排列,取第一个最长的转录本名称tspt_longest_info <- tspt_longest_info[order(tspt_longest_info$width,decreasing = T),]tx_cdsequal_longest_name <- tspt_longest_info$transcript_name[1]# 提取gtf信息储存gene_line <- my_gtf %>% filter(gene_name == g,type == 'gene')cds_txlongest <- my_gtf %>% filter(gene_name == g,transcript_name == tx_cdsequal_longest_name)# 合并gene和转录本信息last_res <- rbind(gene_line,cds_txlongest)# 保存结果到list中longest_gtf[[g]] <- last_res}else{# 如果没有相同cds长度相等的转录本就取cds最长的# 获取最大cds长度max_num <- max(a)# 提取最大最大cds长度对应的转录本名称tn <- data.frame(a)tspt_name <- rownames(tn %>% filter(a == max_num))# 提取gtf信息储存gene_line <- my_gtf %>% filter(gene_name == g,type == 'gene')cds_longest <- my_gtf %>% filter(gene_name == g,transcript_name == tspt_name)# 合并gene和转录本信息last_res <- rbind(gene_line,cds_longest)# 保存结果到list中longest_gtf[[g]] <- last_res}}}
运行结束后,整理查看结果:
# 把结果文件longest_gtf转为data.framemy_longest_gtf <- do.call('rbind',longest_gtf)# 查看基因数量length(table(my_longest_gtf$gene_name))[1] 100# 查转录本数量length(table(my_longest_gtf$transcript_id))[1] 100# 导出 gtf 结果export(my_longest_gtf,'my_longest_transcript.gtf',format = 'gtf')
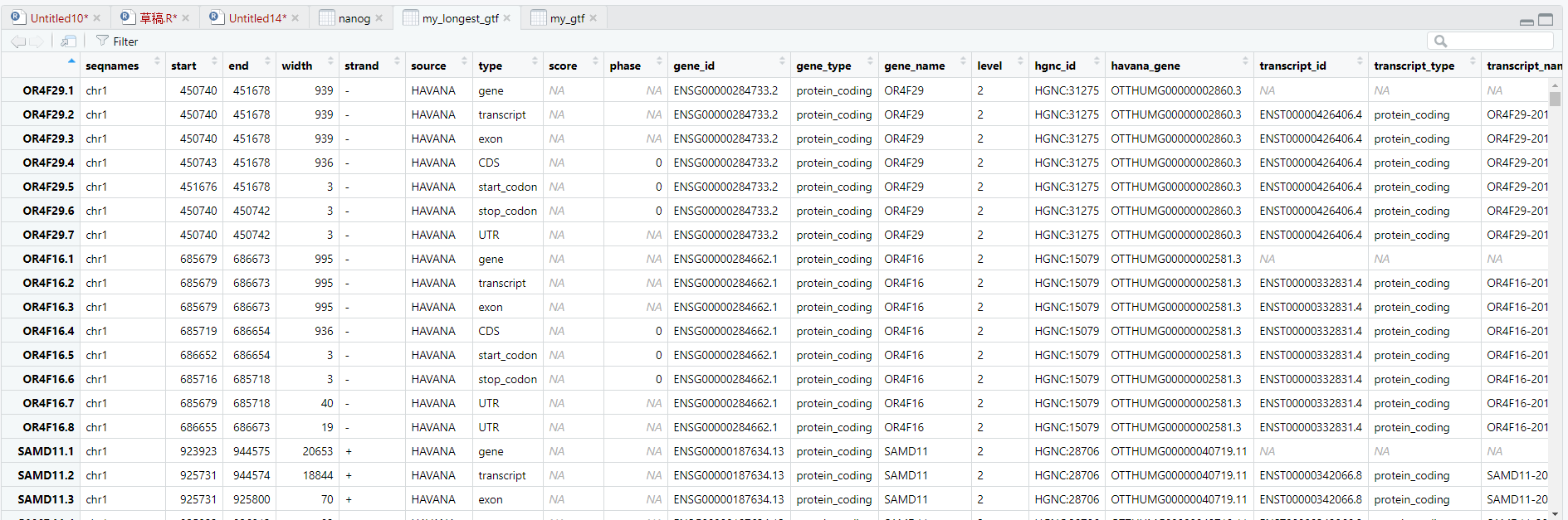
简单验证:
验证 1: 基因有多个转录本,CDS 区只有一个最大值的转录本:
# 挑选基因ginfo <- pt_gtf %>% filter(gene_name == 'NANOG')# 提取转录本tinfo <- ginfo %>% filter(type == 'transcript')# 提取转录本数量ntspt <- nrow(tinfo)ntspt[1] 2# 转录本名称,有两个转录本tname <- tinfo$transcript_nametname[1] "NANOG-201" "NANOG-202"# 提取CDSmtinfo <- ginfo %>% filter(transcript_name == tname[1],type == 'CDS')# 计算cds区总长度,第一个转录本 NANOG-201cds_len <- sum(mtinfo$width)[1] 915mtinfo <- ginfo %>% filter(transcript_name == tname[2],type == 'CDS')# 计算cds区总长度,第二个转录本 NANOG-202cds_len <- sum(mtinfo$width)[1] 867
我们可以看到 NANOG 这个基因有两个转录本NANOG-201、NANOG-202,第一个转录本的 CDS 区比第二个长,我们直接把 NANOG 放到循环里跑一下,只需要把 gene_na 替换成 NANOG 即可,后面代码都是一样:
# 建一个储存结果信息的listlongest_gtf <- list()# 筛选# 测试 NANOG 个基因for (g in 'NANOG'){...
查看筛选的结果:
unique(my_longest_gtf$transcript_name)[1] NA "NANOG-201"
验证 2: 基因有多个转录本,CDS 区有多个最大值的转录本:
# 挑选基因ginfo <- pt_gtf %>% filter(gene_name == 'SMIM1')# 提取转录本tinfo <- ginfo %>% filter(type == 'transcript')# 提取转录本数量ntspt <- nrow(tinfo)ntspt[1] 2# 转录本名称,有两个转录本tname <- tinfo$transcript_nametname[1] "SMIM1-204" "SMIM1-201"# 提取CDSmtinfo <- ginfo %>% filter(transcript_name == tname[1],type == 'CDS')# 计算cds区总长度,第一个转录本 SMIM1-204cds_len <- sum(mtinfo$width)[1] 234mtinfo <- ginfo %>% filter(transcript_name == tname[2],type == 'CDS')# 计算cds区总长度,第二个转录本 SMIM1-201cds_len <- sum(mtinfo$width)[1] 234# 查看转录本长度tinfo %>% filter(transcript_name == tname[1]) %>% select(width)width1 3208tinfo %>% filter(transcript_name == tname[2]) %>% select(width)width1 3196
可以看到 SMIM1 基因有两个转录本 SMIM1-204、SMIM1-201,CDS 长度都一样,转录本长度则是都一个长以一点,我们跑一下循环试试:
# 建一个储存结果信息的listlongest_gtf <- list()# 筛选# 测试 NANOG 个基因for (g in 'SMIM1'){...
查看筛选的结果:
unique(my_longest_gtf$transcript_name)[1] NA "SMIM1-204"
可以看到在 CDS 长度相等的情况下选取了转录本最长的那个。
对转录本 id 修改格式
transcript_id 格式类似于这样 ENST00000642557.4,ensembl 数据库的是没有小数点的,gencode 数据库有,UCSC 数据库的 NM_001385803.1 等类型的,gffread 软件可以提取注释文件转录本序列,将 exon、UTR 等序列合并成完整的 cDNA 序列,输出的序列的名字就是 transcript id,可以在 transcript_id 上多增加一些信息便于后面的筛选或数据分析。
目标
把每个基因转录本的基因名、gene id、transcript id、起始密码子位置、终止密码子位置、转录本长度以下划线连接替换为原来的 transcript_id。
类似于这样:
gene_namegene_idtranscript_idstart_codonstop_codon__transcript_length
分析
本以为比较简单,只要取出每个基因,然后提取出对应的位置信息直接 paste 粘贴在一起,然后重新赋值给 transcript_id 就行了,但后来居然报错了,于是查看了 start codon 和 stop codon 的数量,发现居然比基因或者转录本的数量要多!
也就是说一个转录本有多个起始密码子或者终止密码子,所以赋值的时候长度不一样会报错。
- 注释文件记录的是基因组上的位置!
Start Codon:
画个示意图说明:
从上图我们可以看到,起始密码子中如果没有剪切位点时,记录的位置是连续的,只有一个注释。如果存在剪切位点,记录的位置就不是连续的,会有两个个注释位点的信息。比如像 VAMP3 基因:

VAMP3 基因的起始密码子被截断成了 AT 和第下一个外显子上的第一个碱基 G,中间是内含子部分,所以注释文件会有连个注释信息。这种属于示意图第二个小图里的情况。
Stop Codon:
画个示意图说明:
和 start codon 的情况一样,可以看到,终止密码子中如果没有剪切位点时,记录的位置是连续的,只有一个注释。如果存在剪切位点,记录的位置就不是连续的,会有两个个注释位点的信息。比如像 RPAP2 基因:
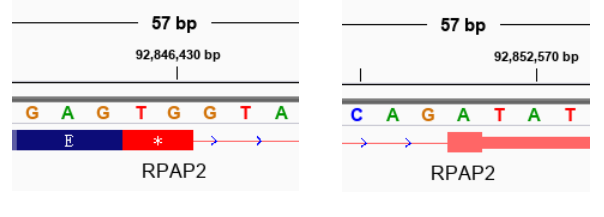
RPAP2 基因的终止密码子被截断成了 TG 和 下一个外显子的 A,中间是内含子部分。这种属于示意图第三个小图里的情况。
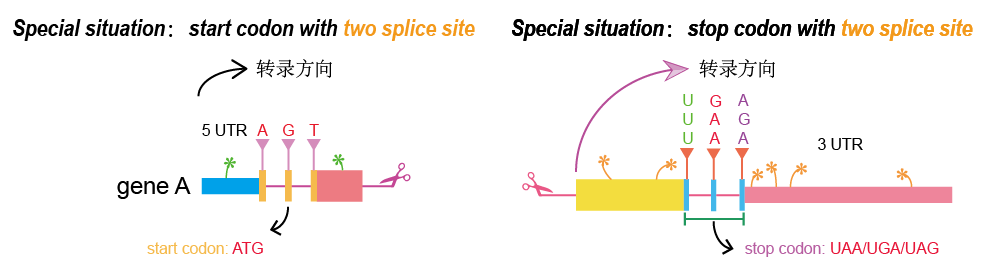
有没有可能有以上这种情况,start 或 stop codon 被两个内含子分开,形成了三个位点,会不会在注释文件里一个转录本有三个 start codon 或 stop codon 信息?这就关系到我上一篇文章里提到的问题了。最短的外显子有一个碱基?
我们可以写个循环判断一下有没有 3 个 start 或 stop codon 注释信息。
思路:
提取每个基因的转录本,循环每个转录本的信息,判断有 1 个、2 个或 3 个 start/stop codon 的数量,符合条件的转录本名字被分别储存在对应的向量里,这样我们就获得了不同起始密码子和终止密码子数量的转录本的数量。
# @@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@@# 判断转录本有几个start codon 或 stop codongene_name <- unique(pt_gtf$gene_name)length(gene_name)[1] 18808# 查看转录本数量length(unique(pt_gtf$transcript_name))[1] 43479# 创建储存含有1、2、3个start codon 和 stop codon的向量# 此外创建没有start codon 或 stop codon的向量t_st1 <- c()t_st2 <- c()t_st3 <- c()no_detected_st <- c()t_sp1 <- c()t_sp2 <- c()t_sp3 <- c()no_detected_sp <- c()
循环代码:
for (g_name in gene_name) {# 提取基因g_dat <- pt_gtf %>% filter(gene_name == g_name)# 提取该基因转录本名称tx_name <- unique(g_dat$transcript_name)# 循环该基因的每个转录本for (tt in tx_name) {# 提取转录本的start codon信息t_st_info <- g_dat %>% filter(transcript_name == tt,type == 'start_codon')# start codon数量st_num <- nrow(t_st_info)# 判断start codon数量,分别储存,分别储存转录本名称if(st_num == 1){t_st1 <- c(tt,t_st1)}else if(st_num == 2){t_st2 <- c(tt,t_st2)}else if(st_num == 3){t_st3 <- c(tt,t_st3)}else{no_detected_st <- c(tt,no_detected_st)}# 提取转录本的stop codon信息t_sp_info <- g_dat %>% filter(transcript_name == tt,type == 'stop_codon')# stop codon数量sp_num <- nrow(t_sp_info)# 判断start codon数量,分别储存转录本名称if(sp_num == 1){t_sp1 <- c(tt,t_sp1)}else if(sp_num == 2){t_sp2 <- c(tt,t_sp2)}else if(sp_num == 3){t_sp3 <- c(tt,t_sp3)}else{no_detected_sp <- c(tt,no_detected_sp)}}}# 查看含有1、2、3个start codon的转录本数量length(t_st1)[1] 43356length(t_st2)[1] 122length(t_st3)[1] 0length(no_detected_st)[1] 1# 查看含有1、2、3个stop codon的转录本数量length(t_sp1)[1] 43390length(t_sp2)[1] 83length(t_sp3)[1] 0length(no_detected_sp)[1] 6
计算出的数量加上正好是所有转录本的数量,侧面证明了代码应该是没有问题的。
由以上结果可以看到,起始密码子或终止密码子是没有 3 个位点的,也就是说最多存在 1 个剪切位点!
此外,我们所选取 start codon 和 stop codon 的位置需要区分正负链,正负链的选取原则是相反的。针对多个起始和终止密码子,如果在正链,取在前面的,负链就取后面的。
下面是改名代码:
# 唯一基因名个数length(unique(my_longest_gtf$gene_name))[1] 100# start codon个数length(my_longest_gtf[which(my_longest_gtf$type == 'start_codon'),'type'])[1] 101# 如VAMP3有两个start codon# stop codon个数length(my_longest_gtf[which(my_longest_gtf$type == 'stop_codon'),'type'])[1] 100# 如RPAP2 基因有两个stop codon# 提取基因名gname <- unique(my_longest_gtf$gene_name)# 建一个储存改名字后的结果信息的listname_changed_gtf <- list()# 循环改名字for (gn in gname) {# 提取基因信息filter_res <- my_longest_gtf %>% filter(gene_name == gn)# gene_namegenename <- gn# gene_idgeneid <- unique(filter_res$gene_id)# transcript_idtsptid <- unique(filter_res$transcript_id)[2]# transcript lengthtspt_length <- filter_res[which(filter_res$type == 'transcript'),'width']# 区分正负链提取start codon 和 stop codon位置信息strand <- unique(filter_res$strand)# 条件语句if (strand == '+') {# 如果在正义链上则取st位点star位置,sp位点取end位置# 如果有多个st或sp密码子,st取最小的,sp取最大的# start codon位置信息# st数量st_num <- length(filter_res[which(filter_res$type == 'start_codon'),'start'])# 循环判断if(st_num == 1){st_pos <- filter_res[which(filter_res$type == 'start_codon'),'start']}else{st_pos <- filter_res[which(filter_res$type == 'start_codon'),'start'][1]}# stop codon位置信息# sp数量sp_num <- length(filter_res[which(filter_res$type == 'stop_codon'),'end'])# 循环判断if(sp_num == 1){sp_pos <- filter_res[which(filter_res$type == 'stop_codon'),'end']}else{sp_pos <- filter_res[which(filter_res$type == 'stop_codon'),'end'][sp_pos]}}else{# 如果在负义链上则取st位点end位置,sp位点取start位置# 如果有多个st或sp密码子,st取最大的,sp最小的# start codon位置信息# st数量st_num <- length(filter_res[which(filter_res$type == 'start_codon'),'end'])# 循环判断if(st_num == 1){st_pos <- filter_res[which(filter_res$type == 'start_codon'),'end']}else{st_pos <- filter_res[which(filter_res$type == 'start_codon'),'end'][st_num]}# stop codon位置信息# sp数量sp_num <- length(filter_res[which(filter_res$type == 'stop_codon'),'start'])# 循环判断if(sp_num == 1){sp_pos <- filter_res[which(filter_res$type == 'stop_codon'),'start']}else{sp_pos <- filter_res[which(filter_res$type == 'stop_codon'),'start'][1]}}# 合并名字:"gene_name"-"gene_id"-"transcript_id"-"start_codon"-"stop_codon"-"transcript_length"comb_name <- paste(genename,geneid,tsptid,st_pos,sp_pos,tspt_length,sep = "_")# 把原来的transcript_id改名filter_res$transcript_id <- comb_name# 结果保存给listname_changed_gtf[[gn]] <- filter_res}# 把结果文件name_changed_gtf转为data.framemy_longest_namechanged_gtf <- do.call('rbind',name_changed_gtf)# 查看结果head(my_longest_namechanged_gtf$transcript_id)[1] "OR4F29_ENSG00000284733.2_ENST00000426406.4_451678_450740_939"[2] "OR4F29_ENSG00000284733.2_ENST00000426406.4_451678_450740_939"[3] "OR4F29_ENSG00000284733.2_ENST00000426406.4_451678_450740_939"[4] "OR4F29_ENSG00000284733.2_ENST00000426406.4_451678_450740_939"[5] "OR4F29_ENSG00000284733.2_ENST00000426406.4_451678_450740_939"[6] "OR4F29_ENSG00000284733.2_ENST00000426406.4_451678_450740_939"# 导出 gtf 结果export(my_longest_namechanged_gtf,'my_longest_namechanged.gtf',format = 'gtf')

若有收获,就点个赞吧
0 人点赞
