做完转录组分析之后,一般都要求做qRT-PCR来验证二代测序得到的转录本表达是否可靠。荧光定量PCR是一种相对表达定量的方法,他的计算方法有很多,常用的相对定量数据分析方法有双标曲线法,ΔCt法,2-ΔΔCt法(Livak法): 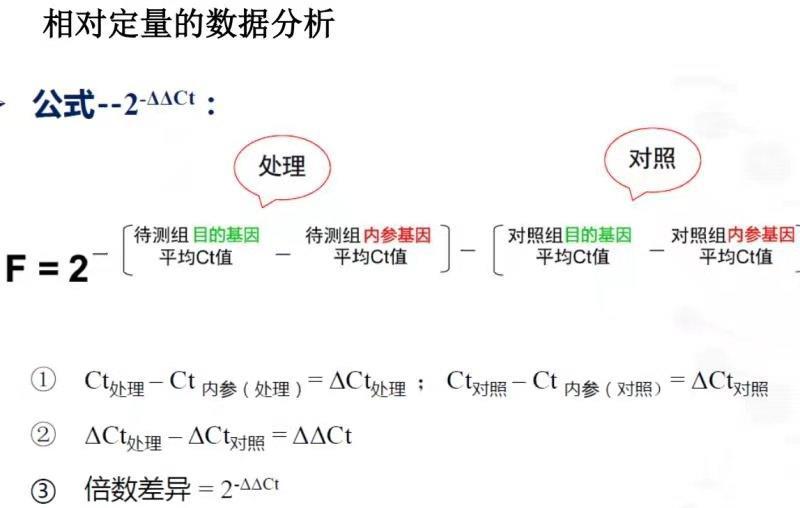 更多知识分享请到 https://zouhua.top/。
更多知识分享请到 https://zouhua.top/。
qRT-PCR介绍及计算公式
该部分引用自下方参考链接1
qRT-PCR原理
以基因的cDNA为模板进行PCR扩增,在PCR扩增过程中,通过收集荧光信号,对PCR进程进行实时检测。由于在PCR扩增的指数时期,模板的Ct值和该模板的起始拷贝数存在线性关系,所以可以定量。
Ct值
Ct值的含义是:每个反应管内的荧光信号达到设定的域值时所经历的循环数 (cycle)。 qRT-PCR在扩增的时候都会有平台期,在平台期之前,PCR 扩增就是简单的指数增长,也就是 1 变 2,2 变 4,4 变 8 …扩增。数学形式就是 2 的 ct 次方,到了平台期所有基因扩增的数目是一致的,而唯一有区别的则是 ct 值的不同。所以不难推断出 ct 值越小,反应扩增到达平台期所需循环数越少,目的基因起始含量越高。这里可以得到公式:
计算 -ΔΔCt:内参基因分为对照组和处理组内参基因
- 先计算对照组和处理组的内参基因Ct的均值: $$Mean_{内参基因}=mean(对照组或处理组内参基因)$$
- 计算对照组待检测目的基因减去对照组内参基因的平均Ct值:$$ΔCt{对照组目的基因i} = Ct{对照组目的基因i} - Ct_{对照组内参基因的平均值}$$
- 计算处理组待检测目的基因减去处理组内参基因的平均Ct值:$$ΔCt{处理组目的基因i} = Ct{处理组目的基因i} - Ct_{处理组内参基因的平均值}$$
- 计算基于对照组的-ΔΔCt,处理组待检测目的基因的ΔCt减去对照组待检测基因的ΔCt的平均值:$$-ΔΔCt{处理组目的基因i} = ΔCt{处理组目的基因i} - ΔCt_{对照组目的基因i的平均值}$$
- 相对表达量计算,也就是相对于对照组: 2^-ΔΔct: $$2^{-(-ΔΔCt)}$$
- 条形图或相关性点图可视化结果
R代码
加载R包
knitr::opts_chunk$set(warning = F, message = F)library(dplyr)library(tibble)library(ggplot2)library(xlsx)library(Rmisc)
R函数
get_qPCR <- function(dataset=dat,ref_gene="GAPDH",control_group="6H NC",grp=c("6H M1")){# dataset=dat # 初始数据# ref_gene="GAPDH" # 内参基因名字# control_group="6H NC" # 对照组# grp=c("6H M1") # 实验组排序if(!any(is.element(colnames(dataset), c("Sample_Name", "Target_Name", "CT")))){stop("Check the sheet's colnames")}sampleid <- c("Sample_Name", "Target_Name", "CT")dat <- dataset %>% select(sampleid)# step1: 计算对照组和处理组的内参基因平均值dat_ref_gene <- dat %>% filter(Target_Name == ref_gene)ref_gene_mean <- dat_ref_gene %>% group_by(Sample_Name) %>%dplyr::summarise(CT_ref_mean = mean(CT))# step2: 计算对照组和处理组待检测目的基因减去对应分组的内参基因的平均Ct值dat_gene <- dat %>% filter(Target_Name != ref_gene)dat_gene_merge <- dat_gene %>% inner_join(ref_gene_mean, by = "Sample_Name")dat_gene_merge$CT_delta <- with(dat_gene_merge, CT - CT_ref_mean)dat_control <- dat_gene_merge %>% filter(Sample_Name == control_group) %>%group_by(Sample_Name, Target_Name) %>%dplyr::summarise(Delta_CT_control_mean=mean(CT_delta)) %>%dplyr::rename(Sample_Name_control=Sample_Name)dat_treat <- dat_gene_merge %>% filter(Sample_Name != control_group) %>%# group_by(Sample_Name, Target_Name) %>%# dplyr::summarise(Delta_CT_treat_mean=mean(CT_delta)) %>%dplyr::rename(Sample_Name_treat=Sample_Name)# step3: 计算对照组检测基因的平均Δ值dat_double_delta <- inner_join(dat_treat, dat_control,by = "Target_Name")dat_double_delta$CT_delta_delta <- with(dat_double_delta, CT_delta - Delta_CT_control_mean)# step4: 基于对照组检测基因的平均Δ值,计算实验组的2-ΔΔCt值dat_double_delta$qPCR <- 2^-(dat_double_delta$CT_delta_delta)# step5: 条形图或相关性散点图可视化dat_plot <- dat_double_delta %>%dplyr::rename(Sample_Name=Sample_Name_treat) %>%dplyr::select(Sample_Name, Target_Name, qPCR)dat_plot_bar <- Rmisc::summarySE(dat_plot, measurevar = "qPCR",groupvars = c("Sample_Name", "Target_Name")) %>%mutate(Sample_Name=factor(Sample_Name, levels = grp),Target_Name=factor(Target_Name)) %>%group_by(Sample_Name, Target_Name) %>%mutate(ylimit=(qPCR+sd)) %>%ungroup()dat_plot_bar_ymax <- dat_plot_bar %>%group_by(Target_Name) %>%summarise_at(vars(ylimit), max)# dat_plot_range <- dat_plot %>% group_by(Sample_Name, Target_Name) %>%# summarise(ymin=min(qPCR), ymax=max(qPCR))# setting y axis scaley_group <- c()y_scale <- c()for(i in 1:nrow(dat_plot_bar_ymax)){y_group <- c(y_group, rep(as.character(dat_plot_bar_ymax$Target_Name[i]), 2))y_scale <- c(y_scale, c(0, ceiling(dat_plot_bar_ymax$ylimit[i])))}blank_data <- data.frame(Target_Name = y_group,Sample_Name = 1,qPCR = y_scale)# step6: visualizationpl <- ggplot(dat_plot_bar, aes(x=Sample_Name, weight=qPCR))+geom_hline(aes(yintercept = qPCR), color = "gray")+geom_bar(color = "black", width = .4, position = "dodge")+geom_errorbar(aes(ymin = qPCR, ymax = qPCR + se),width = 0.25, size = 0.5, position = position_dodge(0.7))+labs(x="", y=expression(paste(log[2], " fold change in expression")))+geom_blank(data = blank_data, aes(x = Sample_Name, y = qPCR))+expand_limits(y = 0)+scale_y_continuous(expand = c(0, 0))+facet_wrap(. ~ Target_Name, scales = "free")+theme_bw()+theme(axis.title = element_text(face = "bold", color = "black", size = 14),axis.text = element_text(color = "black", size = 10),axis.text.x = element_text(angle = 60, hjust = 1, face = "bold"),text = element_text(size = 10, color = "black", family="serif"),panel.grid = element_blank(),legend.position = "right",legend.key.height = unit(0.6, "cm"),legend.text = element_text(face = "bold", color = "black", size = 10),strip.text = element_text(face = "bold", size = 14))res <- list(dat=dat_double_delta, plot=pl)return(res)}
读取数据
单个样本三个技术重复,检验不同的目的基因扩增效率
dat <- read.xlsx("qPCR.xlsx", sheetIndex = 1)head(dat)

计算结果
qPCR_res <- get_qPCR(dataset=dat,ref_gene="GAPDH",control_group="6H NC",grp=c("6H M1"))DT::datatable(qPCR_res$dat)

可视化结果
qPCR_res$plot

结果: IL-1B 和INOS基因相比NC组而言,其含量越多
同一基因多分组结果图
get_qPCR <- function(dataset=dat,ref_gene="Gadph",control_group="2d_Control",grp=c("2d_100uM")){# dataset=dat # 初始数据# ref_gene="Gadph" # 内参基因名字# control_group="2d_Control" # 对照组# grp=c("2d_100uM") # 实验组排序if(!any(is.element(colnames(dataset), c("Sample_Name", "Target_Name", "CT")))){stop("Check the sheet's colnames")}sampleid <- c("Sample_Name", "Target_Name", "CT")dat <- dataset %>% select(sampleid)# step1: 计算对照组和处理组的内参基因平均值dat_ref_gene <- dat %>% filter(Target_Name == ref_gene)ref_gene_mean <- dat_ref_gene %>% group_by(Sample_Name) %>%dplyr::summarise(CT_ref_mean = mean(CT))# step2: 计算对照组和处理组待检测目的基因减去对应分组的内参基因的平均Ct值dat_gene <- dat %>% filter(Target_Name != ref_gene)dat_gene_merge <- dat_gene %>% inner_join(ref_gene_mean, by = "Sample_Name")dat_gene_merge$CT_delta <- with(dat_gene_merge, CT - CT_ref_mean)dat_control <- dat_gene_merge %>% filter(Sample_Name == control_group) %>%group_by(Sample_Name, Target_Name) %>%dplyr::summarise(Delta_CT_control_mean=mean(CT_delta)) %>%dplyr::rename(Sample_Name_control=Sample_Name)dat_treat <- dat_gene_merge %>% filter(Sample_Name != control_group) %>%# group_by(Sample_Name, Target_Name) %>%# dplyr::summarise(Delta_CT_treat_mean=mean(CT_delta)) %>%dplyr::rename(Sample_Name_treat=Sample_Name)# step3: 计算对照组检测基因的平均Δ值dat_double_delta <- inner_join(dat_treat, dat_control,by = "Target_Name")dat_double_delta$CT_delta_delta <- with(dat_double_delta, CT_delta - Delta_CT_control_mean)# step4: 基于对照组检测基因的平均Δ值,计算实验组的2-ΔΔCt值dat_double_delta$qPCR <- 2^-(dat_double_delta$CT_delta_delta)# step5: 条形图或相关性散点图可视化dat_plot <- dat_double_delta %>%dplyr::rename(Sample_Name=Sample_Name_treat) %>%dplyr::select(Sample_Name, Target_Name, qPCR)dat_plot_bar <- Rmisc::summarySE(dat_plot, measurevar = "qPCR",groupvars = c("Sample_Name", "Target_Name")) %>%mutate(Sample_Name=factor(Sample_Name, levels = grp))# step6: visualizationpl <- ggplot(dat_plot_bar, aes(x=Sample_Name, weight=qPCR))+geom_hline(yintercept = seq(0, round(max(dat_plot_bar$qPCR), 1), 0.2), color = "gray")+geom_bar(color = "black", width = .4, position = "dodge")+geom_errorbar(aes(ymin = qPCR, ymax = qPCR + se),width = 0.25, size = 0.5, position = position_dodge(0.7))+labs(x="", y=expression(paste(log[2], " fold change in expression")))+scale_y_continuous(breaks = seq(0, round(max(dat_plot_bar$qPCR), 1), 0.2),expand = c(0, 0),limits = c(0, round(max(dat_plot_bar$qPCR), 1)+round(max(dat_plot_bar$sd), 1)))+facet_wrap(. ~ Target_Name, scales = "free")+theme_bw()+theme(axis.title = element_text(face = "bold", color = "black", size = 14),axis.text = element_text(color = "black", size = 10),axis.text.x = element_text(angle = 60, hjust = 1, face = "bold"),text = element_text(size = 10, color = "black", family="serif"),panel.grid = element_blank(),legend.position = "right",legend.key.height = unit(0.6, "cm"),legend.text = element_text(face = "bold", color = "black", size = 10),strip.text = element_text(face = "bold", size = 14))res <- list(dat=dat_double_delta, plot=pl)return(res)}dat <- read.xlsx("qPCR.xlsx", sheetName = "6d")# 6 daysqPCR_res <- get_qPCR(dataset=dat,ref_gene="Gapdh",control_group="6d_control",grp=c("6d_100", "6d_300", "6d_1000"))# 计算结果DT::datatable(qPCR_res$dat)# 可视化结果qPCR_res$plot

参考
参考文章如引起任何侵权问题,可以与我联系,谢谢。

若有收获,就点个赞吧
0 人点赞
