首先推荐的资料,下面分享的相关知识,主要都是从这些资料中获取的
Current best practices in single‐cell RNA‐seq analysis: a tutorial
单细胞相关教程
分析流程:(以10x单细胞测序数据为例)
大致可分为:
1.fastq转换为raw data
2.数据质控QC
3.数据标准化,归一化处理
4.数据整合,去除生物偏差(细胞周期)和技术偏差(批次效应)
5.聚类cluster
6.cluster的marker gene识别和cluster的注释, DEG
7.细胞的轨迹分析
1.fastq转换为raw data
对于10x单细胞数据而言,可以使用cell ranger官网软件对获取的fastq数据进行处理。
详细使用介绍可参考10X单细胞测序分析软件:Cell ranger
cell ranger处理流程分为:数据拆分(cellranger mkfastq)、细胞定量(cellranger count)、组合分析(cellranger aggr)、参数调整(cellranger reanalyze)
1.1 数据拆分
cellranger mkfastq中封装了Illumina’s bcl2fastq软件,用来拆分Illumina 原始数据(raw base call (BCL)),输出 FASTQ 文件。
cellranger mkfastq --id=tiny-bcl \--run=/path/to/tiny_bcl \--csv=cellranger-tiny-bcl-simple-1.2.0.csvcellranger mkfastq --id=tiny-bcl \--run=/path/to/tiny_bcl \--samplesheet=cellranger-tiny-bcl-samplesheet-1.2.0.csv
1.2 细胞定量
cellranger count 完成细胞和基因的定量,也就是产生了我们用来做各种分析的基因表达矩阵。
cellranger count --id=run_count_1kpbmcs \--fastqs=/run_cellranger_count/pbmc_1k_v3_fastqs \--sample=pbmc_1k_v3 \--transcriptome=/run_cellranger_count/refdata-cellranger-GRCh38-3.0.0
id:对你运行的项目起个名字,可以任意取名(输出结果在建文件夹时以这个名字命名)
fastqs:包含fastq文件的路径
sample:如果上述路径中包含的文件不只一个样本的,则需要指定该参数,该参数是根据fastq文件名的前缀对文件进行识别的,可以用来区分不同的样本
transcriptome:用来保存参考基因组的路径
.outs├── analysis【数据分析文件夹】│ ├── clustering【聚类,图聚类和k-means聚类】│ ├── diffexp【差异分析】│ ├── pca【主成分分析线性降维】│ └── tsne【非线性降维信息】├── cloupe.cloupe【Loupe Cell Browser 输入文件】├── filtered_feature_bc_matrix【过滤后的barcode信息,是后续分析的输入文件】│ ├── barcodes.tsv.gz│ ├── features.tsv.gz│ └── matrix.mtx.gz├── filtered_feature_bc_matrix.h5【过滤后的barcode信息HDF5 format】├── metrics_summary.csv【CSV format数据摘要】├── molecule_info.h5【UMI信息,aggregate的时候会用到的文件】├── raw_feature_bc_matrix【原始barcode信息】│ ├── barcodes.tsv.gz│ ├── features.tsv.gz│ └── matrix.mtx.gz├── possorted_genome_bam.bam【比对文件】├── possorted_genome_bam.bam.bai【索引文件】├── raw_feature_bc_matrix.h5【原始barcode信息HDF5 format】├── web_summary.html【网页简版报告以及可视化】└── *_gene_bar.csv_temp【过程文件】
1.3 组合分析
cellranger aggr 对多个GEM的结果进行合并
When doing large studies involving multiple GEM wells, run cellranger count on FASTQ data from each of the GEM wells individually, and then pool the results using cellranger aggr, as described here
可以将同组的不同测序样本的表达矩阵进行合并,整合在一起。
如:toumor组有4个样本,合并在一起,最后生成一个toumor的矩阵
cd /home/jdoe/runscellranger aggr --id=AGG123 \--csv=AGG123_libraries.csv \--normalize=mapped
library_id,molecule_h5LV123,/opt/runs/LV123/outs/molecule_info.h5LB456,/opt/runs/LB456/outs/molecule_info.h5LP789,/opt/runs/LP789/outs/molecule_info.h5library_id,molecule_h5,batchLV123,/opt/runs/LV123/outs/molecule_info.h5,v2_libLB456,/opt/runs/LB456/outs/molecule_info.h5,v3_libLP789,/opt/runs/LP789/outs/molecule_info.h5,v3_lib
8.细胞类型的注释
可以使用SingleR和cellassign的两种方法对各cluster的细胞类型进行注释。
SingleR
https://bioconductor.org/packages/release/bioc/html/SingleR.html
https://github.com/dviraran/SingleR
cellassign
scCATCH
https://github.com/ZJUFanLab/scCATCH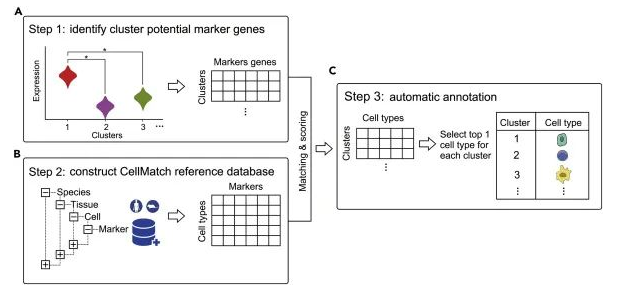
特点:
- CellMatch包含353种细胞类型和686种亚型,与184种组织类型,20,792种细胞特异性标记基因以及2,097个人类和小鼠参考文献。
- scCATCH主要包括两个函数
“findmarkergenes”和“scCATCH”,以实现对每个已识别集群的自动注释。 - scCATCH可用于注释癌组织的scRNA-seq数据。
- scCATCH可以处理包含超过10,000个细胞和15个以上clusters的大型单细胞转录组数据集。
Note:
(1)只能注释人或小鼠;
(2)在数据库中,人的组织和肿瘤以及小鼠的正常组织的参比资源很多,小鼠的肿瘤组织较少。
_
手动注释
https://www.jianshu.com/p/0d5c546b823e

若有收获,就点个赞吧
0 人点赞
