1.0 准备工作
安装Python > 3.7
jupyter 代码运行客户端
scanpy 分析单细胞数据
seaborn 可视化数据结果
stream 轨迹分析
GSEAPY GO/KEGG富集分析
Squidpy 空间转录组分析、细胞互作分析
1.1 导入安装包
import scanpy as scimport numpy as npimport pandas as pdimport matplotlib.pyplot as pltimport seaborn as snsfrom statannot import add_stat_annotation%matplotlib inline%config InlineBackend.figure_format = 'svg'warnings.filterwarnings("ignore")sc.settings.verbosity = 3 # 设置日志等级: errors (0), warnings (1), info (2), hints (3)sc.logging.print_header()sc.settings.set_figure_params(dpi=80, facecolor='white')
1.2 读入数据
案例都是以下载数据集。本文导入自己10X下机数据,一个肺癌样本 分别是Tumor 和 Normal
adata_T = sc.read_10x_mtx("/data/public/scRNA/data/lyk/3sample_Lung/Tumor_filtered_feature_bc_matrix",var_names='gene_symbols',cache=True)adata_N = sc.read_10x_mtx("/data/public/scRNA/data/lyk/3sample_Lung/Normal_filtered_feature_bc_matrix/",var_names='gene_symbols',cache=True)
1.3 数据处理
- 去除重复基因 ```python adata_T.var_names_make_unique() adata_N.var_names_make_unique()
- 合并数据```pythonadata_all = adata_T.concatenate(adata_N, batch_categories=['T',"N"])
- 查看数据
adata_all

obs 为细胞维度对象 日后细胞操作均在osb对象中操作,var是基因维度对象,同理基因在var对象。初始数据仅两个对象。后面计算将慢慢添加。
自定义名称
adata_all.obs['sample'] = adata_all.obs['batch']
高表达基因
sc.pl.highest_expr_genes(adata_all, n_top=20, )
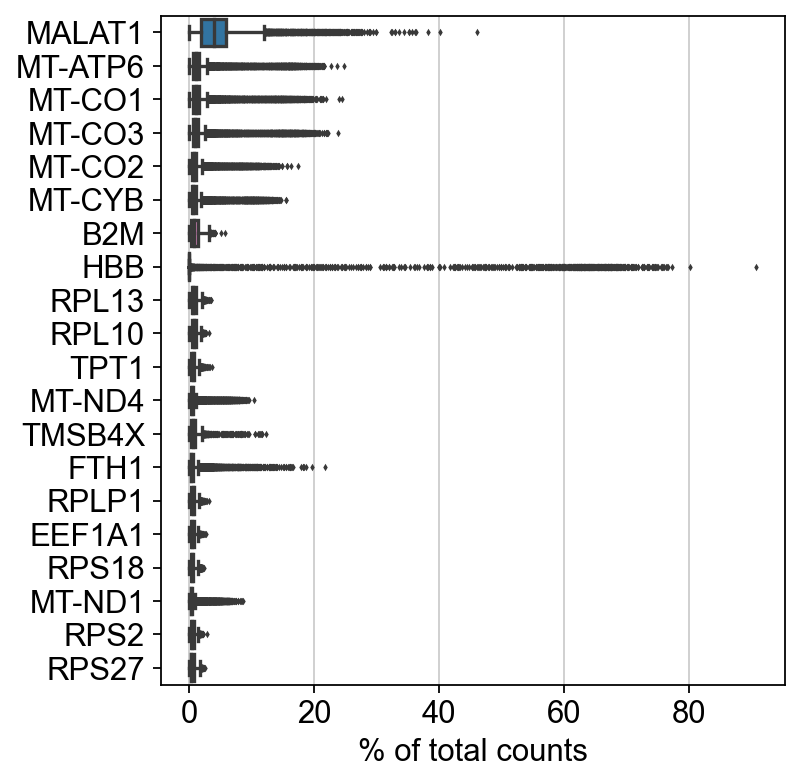
高表达基因 大多数是 线粒体等基因 对以后降维聚类有影响 下一步过滤线粒体数据。
数据过滤
sc.pp.filter_cells(adata_all, min_genes=500) # 去除表达基因500以下的细胞sc.pp.filter_genes(adata_all, min_cells=50) # 去除在100个细胞以下表达的基因sc.pp.filter_cells(adata_all, max_genes = 5000) # 去除基因数超过5000的细胞sc.pp.filter_cells(adata_all, min_counts= 1000) # 去除umi数超过1000的细胞sc.pp.filter_cells(adata_all, max_counts= 25000)# 去除umi数超过25000的细胞
过滤标准 依据数据量而定。
计算MT含量 并写入obs对象
mito_genes = adata_all.var_names.str.startswith('MT-')adata_all.obs['percent_mito'] = np.sum(adata_all[:, mito_genes].X, axis=1).A1 / np.sum(adata_all.X, axis=1).A1adata_all.obs['n_counts'] = adata_all.X.sum(axis=1).A1adata_all

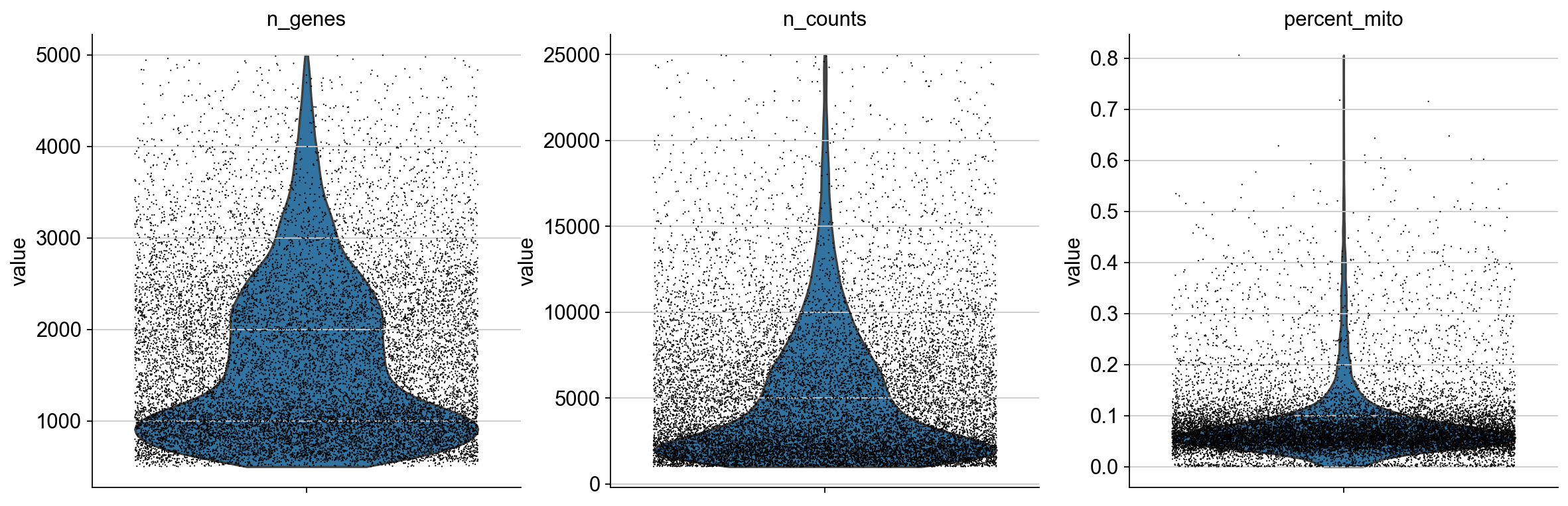
质控图示
sc.pl.violin(adata_all, ['n_genes', 'n_counts', 'percent_mito'],jitter=0.4, multi_panel=True)
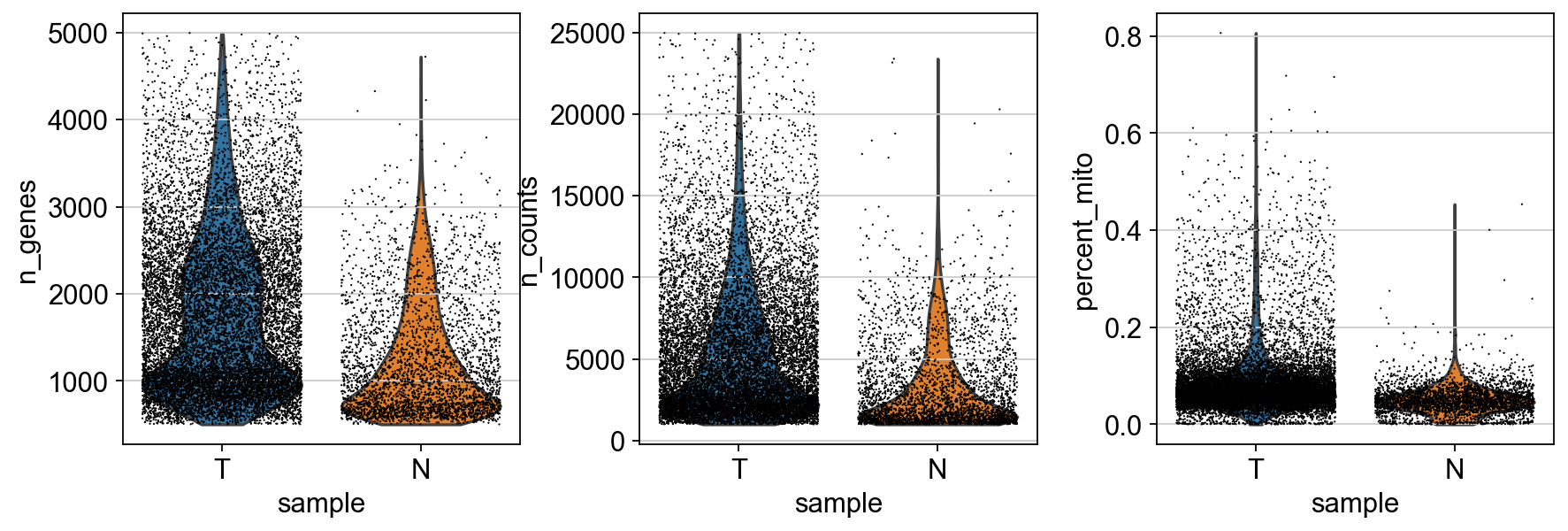
#按照样本画图sc.pl.violin(adata_all, ['n_genes', 'n_counts', 'percent_mito'],jitter=0.4, multi_panel=True,groupby="sample")
# 散点图 形式展示线粒体和gene数目 和umi 关系sc.pl.scatter(adata_all, x='n_counts', y='percent_mito')sc.pl.scatter(adata_all, x='n_counts', y='n_genes')
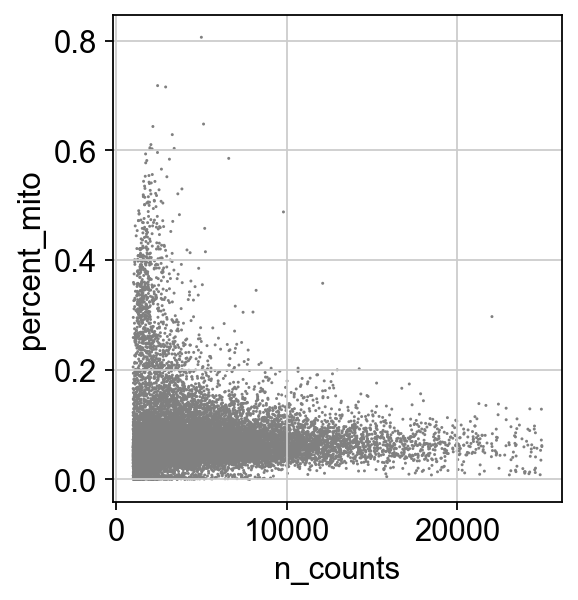
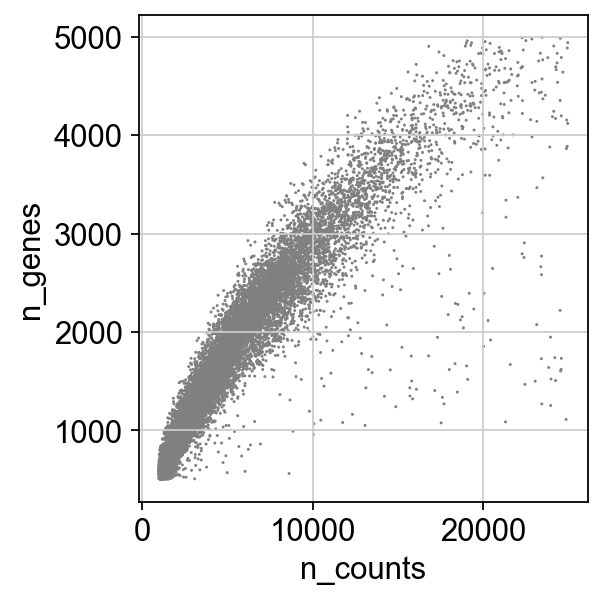
过滤高比例线粒体细胞
adata_all = adata_all[adata_all.obs.pct_counts_mt < 10, :] # 可自行画图查看过滤前后数据
计算双细胞
sc.external.pp.scrublet(adata_all, expected_doublet_rate = 0.05, threshold = 0.25)adata_all.obs.groupby('sample')['doublet_score'].describe()
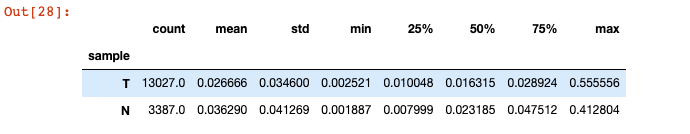
过滤双细胞
adata_all = adata_all[adata_all.obs.predicted_doublet == False, : ]mask = ( adata_all.obs['predicted_doublet'] == False) & (adata_all.obs.doublet_score < 0.25)mask.value_counts()adata_all = adata_all[ mask, :]adata_all

obs 添加了n_genes n_conuts percent_mito sample doublet_score predicted_doublet 等细胞维度数据。uns 新一个对象添加了各总类计算分数结果。
到此为止 数据质控基本完毕。


若有收获,就点个赞吧
0 人点赞
