5.1 导读
后续高级分析不乏有之前画图方式,所有可视化画图均和数据以及参数有关系。高级分析分为以下几种:
- 细胞互作分析
- 差异基因分析
- GO/KEGG 注释
- 细胞轨迹(伪时间序列)
5.2 细胞互作分析
细胞互作分析 常用的cellphone DB 但是这个库没有API 可以直接调用,不过在GitHub上有人引用了cellphone DB 数据库做成了API可以在jupyter里面直接写代码跑数据。
import squidpy as sq # 该包是专门做空间转录组数据的res = sq.gr.ligrec(adata_all,n_perms=1000,cluster_key="cell_type",copy=True,use_raw=False,transmitter_params={"categories": "ligand"},receiver_params={"categories": "receptor"},interactions_params={'resources': 'CellPhoneDB'})
引用受体和配体库,
CellPhoneDB。cluster_key参数为选择互作的类型,如果有特殊需求可以进行筛选数据进行分析。比如B细胞和别的细胞互作,可进行筛选数据进行分析。
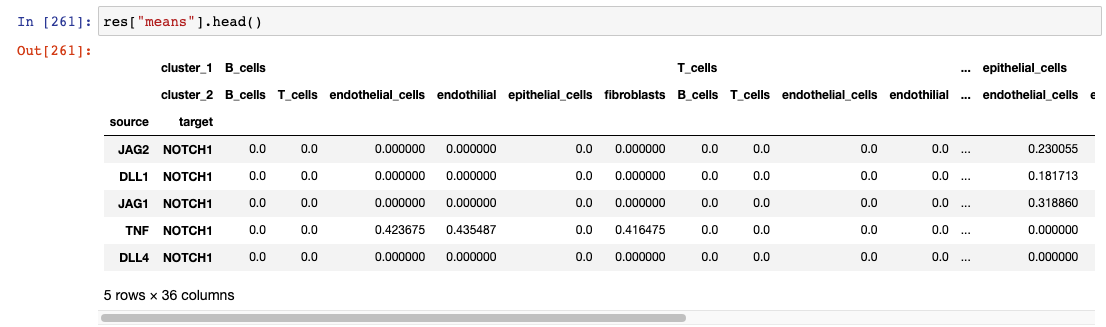
sq.pl.ligrec(res, source_groups=['B_cells',"T_cells"],target_groups=['endothelial_cells',"epithelial_cells","fibroblasts"] ,alpha=0.0005,swap_axes=True,figsize=(30,5))
5.3 差异基因分析
差异基因分析 可以选择数据进行分析,本案例选择T 样本中B细胞和 N样本中的B细胞进行差异分析。
筛选数据
adata_B = adata_all[adata_all.obs["cell_type"].isin(["B_cells"])]## adata_B 仅有B细胞在里面sc.tl.rank_genes_groups(adata_B, groupby='sample', reference="N",method='wilcoxon', n_genes =None, pts = True,key_added="rank_gene_B" )## 用N做对照进行样本之间分析差异基因 数据存入`rank_gene_B`中
提取差异基因进行查看
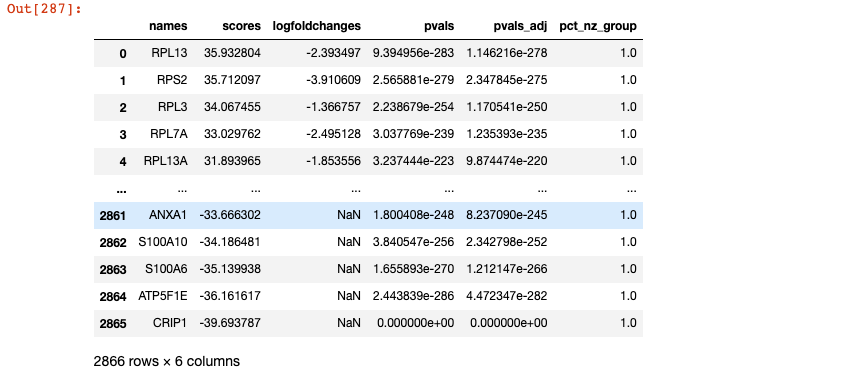
dedf = sc.get.rank_genes_groups_df(adata_B,group=None, pval_cutoff =0.05, log2fc_min=None)dedf
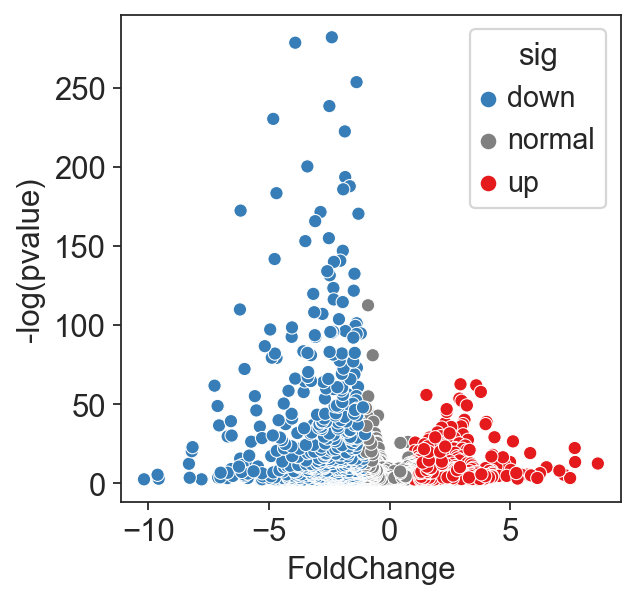
火山图可视化 ```python dedf[‘log(pvalue)’] = -np.log10(dedf[‘pvals’])
dedf[‘sig’] = ‘normal’
dedf[‘size’] =np.abs(dedf[‘logfoldchanges’])/10
dedf.loc[(dedf.logfoldchanges> 1 )&(dedf.pvals < 0.05),’sig’] = ‘up’ dedf.loc[(dedf.logfoldchanges< -1 )&(dedf.pvals < 0.05),’sig’] = ‘down’ ax = sns.scatterplot(x=”logfoldchanges”, y=”log(pvalue)”, hue=’sig’, hue_order = (‘down’,’normal’,’up’), palette=(“#377EB8”,”grey”,”#E41A1C”), data=result) ax.set_ylabel(‘-log(pvalue)’,fontweight=’bold’) ax.set_xlabel(‘FoldChange’,fontweight=’bold’)
<a name="tlV4n"></a>#### 5.4 GO/KEGG 注释> 利用上述的差异基因进行GO/KEGG 注释```pythonimport gseapy as gpnames = gp.get_library_name() # default: Humannames[:10]
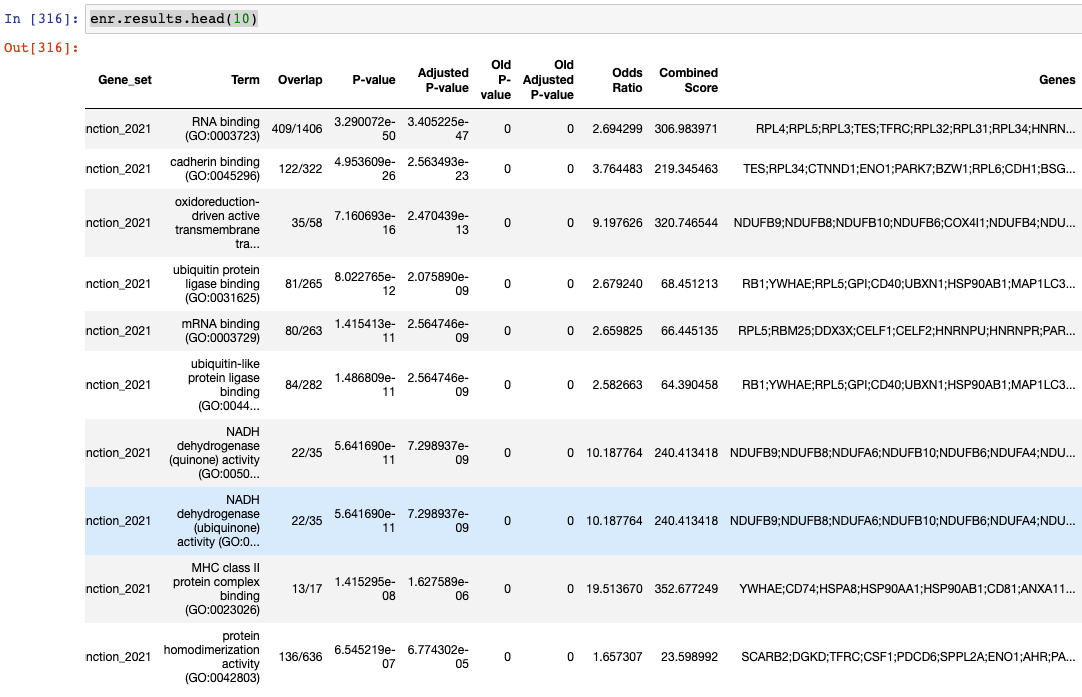
gene_list = list(dedf['names']) # 获取上述差异基因列表enr = gp.enrichr(gene_list=gene_list,gene_sets=['GO_Molecular_Function_2021',"GO_Cellular_Component_2021","GO_Biological_Process_2021"],organism='Human',description='test_name',outdir='test/enrichr_kegg',no_plot=True,cutoff=0.05 # test dataset, use lower value from range(0,1))enr.results.head(10)
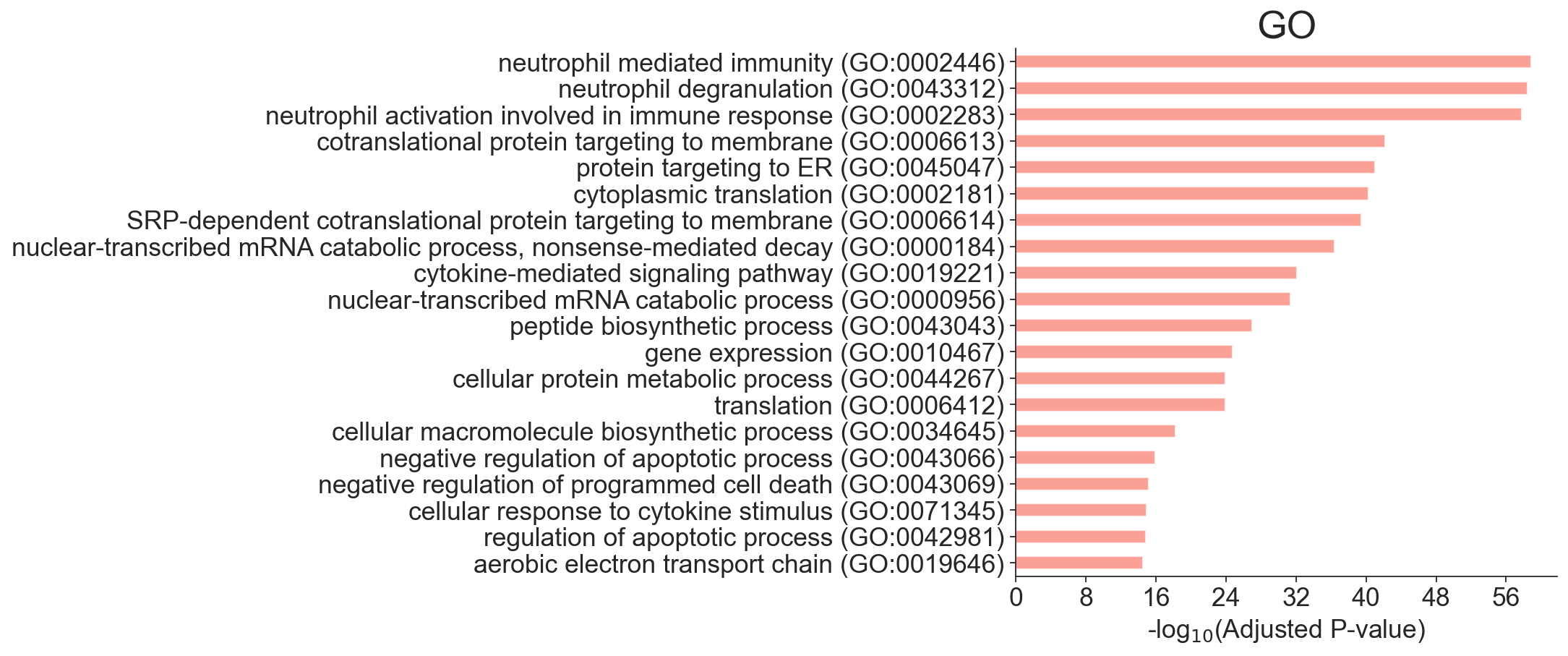
- 可视化GO
ax = barplot(enr.res2d,cutoff=0.05,title='GO',top_term=20,figsize=(16,16))
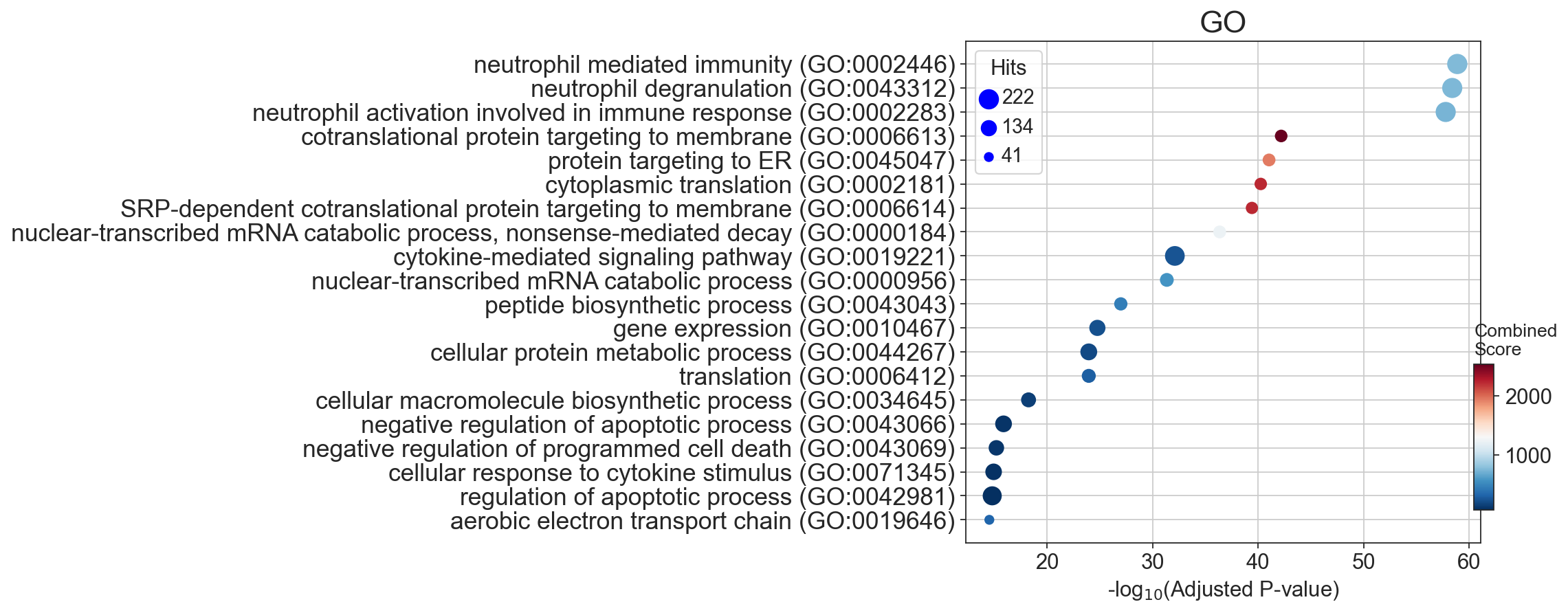
ax = dotplot(enr.res2d,cutoff=0.05,title='GO',top_term=20,figsize=(6,6))
KEGG 同样的流程,如果觉得图不好看 可以用结果数据自行画图。
细胞轨迹分析
细胞轨迹 两种方法。一种是scanpy 自带分析。一种
**stream**。
- fa计算轨迹图 ```python sc.tl.draw_graph(adata_all) sc.pl.draw_graph(adata_all, color=’cell_type’, legend_loc=’on data’)
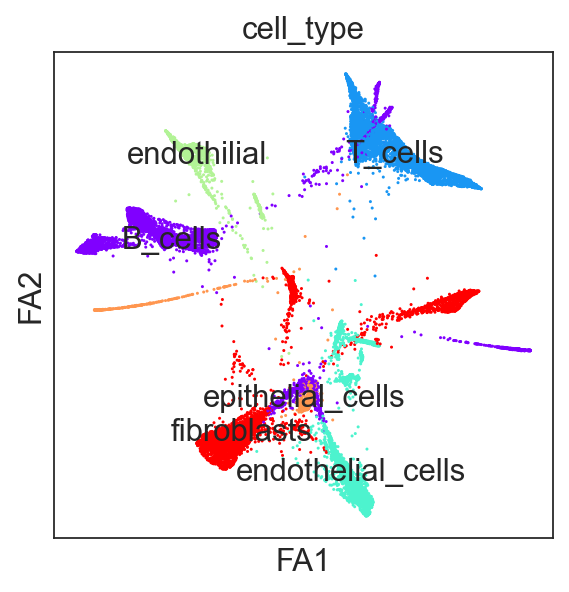- paga 计算轨迹图```pythonsc.tl.paga(adata_all, groups='cell_type')sc.settings.set_figure_params(dpi=150, facecolor='white',dpi_save =100)sc.pl.paga(adata_all, color=['cell_type'],fontsize=5)

**stream**运用https://github.com/pinellolab/STREAM 对系统gcc有要求 stream 有自己导入数据格式,目前还没找到和scanpy互通的API以后专门为STREAM写文档。 有兴趣的可以自己研究一下。


若有收获,就点个赞吧
0 人点赞
