- Single-cell RNA-seq clustering analysis: aligning cells across conditions
- Normalization, variance stabilization, and regression of unwanted variation for each sample
- Integrate samples using shared highly variable genes
- UMAP的可视化
zhuang xioajin
6月-24-2021
Single-cell RNA-seq clustering analysis: aligning cells across conditions
- 准确 normalize and scale the gene expression values,以解决测序深度和过度分散计数值的差异。
- 为了 找出最变异基因可能成为指示细胞类型呈现不同的。
- To align similar cells across conditions.
挑战:
- Checking and removing unwanted variation so that we do not have cells clustering by artifacts downstream
- Aligning cells of similar cell types so that we do not have cell-type specific clustering downstream
建议:
- 在执行聚类之前,请充分了解您对存在的细胞类型的期望。了解您是否期望低复杂性或更高线粒体含量的细胞类型以及细胞是否正在分化
- 如果需要且适合实验, Regress out UMI 的数量(默认使用 sctransform)、线粒体含量和细胞周期,因此不会推动下游聚类
为了识别集群,将执行以下步骤:
- 每个样本的 Normalization、 variance stabilization, and regression of unwanted variation (例如线粒体转录本丰度、细胞周期阶段等)的回归
- 使用共享的高度可变基因整合样本(可选,但如果细胞类型按样本/条件分离,则建议对齐来自不同样本/条件的细胞)
- 基于顶级PC(metagenes)的 Clustering cells
- 质量控制指标探索:确定簇是否与 UMI、基因、细胞周期、线粒体含量、样本等不平衡。
- 使用 known cell type-specific gene markers 搜索预期的细胞类型
# Single-cell RNA-seq analysis - clustering analysis# Load librariesrm(list = ls())library(Seurat)library(tidyverse)library(RCurl)library(cowplot)## 加载筛选过的数据load("data/seurat_filtered.RData")
1. Normalization, variance stabilization, and regression of unwanted variation for each sample
分析的第一步是对原始计数进行归一化,以考虑每个样品每个细胞的测序深度的差异。Seurat最近推出了一种新的方法,用于scRNA-seq数据的归一化和方差稳定化,称为 sctransform。
sctransform方法使用正则化的负二项式模型对UMI计数进行建模,以消除由于测序深度(每个细胞的总nUMIs)引起的变化,同时根据具有类似丰度的基因之间的集合信息来调整方差(类似于一些批量RNA-seq方法)。
output of the model (残差)是测试的每个转录本的归一化表达水平。
Sctransform自动回归出测序深度(nUMIs);然而,数据中还有其他无趣的变化来源,这些变化往往是针对数据集的。例如,对于一些数据集,细胞周期阶段可能是一个重要的变化来源,而对于其他数据集则不是。在你将回归出由于细胞周期阶段引起的变化之前,你需要检查细胞周期阶段是否是数据中变化的主要来源。
1.1 Cell cycle scoring
建议在执行sctransform方法前检查细胞周期阶段。由于细胞之间的计数需要具有可比性,而每个细胞的UMI总数不同,我们通过除以每个细胞的总计数并取自然对数来做一个粗略的归一化。这种方法不如我们最终用来识别细胞集群的sctransform方法准确,但它足以探索我们数据中的变化来源。
# Normalize the counts
seurat_phase <- NormalizeData(filtered_seurat)
一旦数据被归一化为测序深度,我们就可以根据每个细胞的G2/M和S期标志物的表达情况给它打分。
# Load cell cycle markers
lns <- load("data/cycle.rda")
lns
## [1] "g2m_genes" "s_genes"
# Score cells for cell cycle
seurat_phase <- CellCycleScoring(seurat_phase,
g2m.features = g2m_genes,
s.features = s_genes)
# View cell cycle scores and phases assigned to cells
View(seurat_phase@meta.data)
在对细胞周期进行评分后,我们想用PCA来确定细胞周期是否是我们数据集中变化的一个主要来源。为了进行PCA,我们需要首先选择变化最大的特征,然后对数据进行缩放。由于高表达的基因表现出最高的变异量,而我们不希望我们的 “高变异基因”只反映高表达,所以我们需要对数据进行缩放,使变异与表达水平成比例。Seurat ScaleData()函数将通过以下方式对数据进行缩放。
- 调整每个基因的表达,使整个细胞的平均表达量为0
- 按比例调整每个基因的表达量,使各细胞的方差为1
# Identify the most variable genes
seurat_phase <- FindVariableFeatures(seurat_phase,
selection.method = "vst",
nfeatures = 2000,
verbose = FALSE)
# Scale the counts
seurat_phase <- ScaleData(seurat_phase)
注意:对于selection.method和nfeatures参数,指定的值是默认设置。因此,你不一定需要在你的代码中包括这些。我们把它包括在这里是为了提高透明度,并告知你正在使用什么。
# Perform PCA
seurat_phase <- RunPCA(seurat_phase)
# Plot the PCA colored by cell cycle phase
DimPlot(seurat_phase,
reduction = "pca",
group.by= "Phase",
split.by = "Phase")

我们没有看到由于细胞周期阶段而产生的巨大差异。根据这个图,我们不会将细胞周期引起的变化回归出来。
1.2 SCTransform
现在我们可以使用sctransform方法作为一种更准确的归一化方法,估计原始过滤数据的方差,并确定最易变的基因。默认情况下,sctransform考虑了细胞测序深度,或nUMIs。
我们已经检查了细胞周期,并决定它不代表我们数据中的主要变异来源,但线粒体表达是另一个可以大大影响聚类的因素。很多时候,将线粒体表达引起的变异regressing出来是很有用的。然而,如果线粒体基因表达的差异代表了一种生物现象,可能有助于区分细胞集群,那么我们建议不要对线粒体表达进行regressing。
我们可以使用 “for loop”对每个样本运行NormalizeData()、CellCycleScoring()和 SCTransform(),并通过在SCTransform()函数的 vars.to.regress参数中指定regress出线粒体表达。
在我们运行这个for循环之前,我们知道在内存方面,输出可以产生大的R对象/变量。如果我们有一个大的数据集,那么我们可能需要使用以下代码调整R内部允许的对象大小的限制(默认是500 * 1024 ^ 2 = 500 Mb)。
options(future.globals.maxSize = 4000 * 1024^2)
现在,对所有样品进行细胞周期评分和sctransform。这可能需要一些时间(~10分钟)。
# Split seurat object by condition to perform cell cycle scoring and SCT on all samples
split_seurat <- SplitObject(filtered_seurat, split.by = "sample")
split_seurat <- split_seurat[c("ctrl", "stim")]
for (i in 1:length(split_seurat)) {
split_seurat[[i]] <- NormalizeData(split_seurat[[i]], verbose = TRUE)
split_seurat[[i]] <- CellCycleScoring(split_seurat[[i]], g2m.features=g2m_genes, s.features=s_genes)
split_seurat[[i]] <- SCTransform(split_seurat[[i]], vars.to.regress = c("mitoRatio"))
}
##
|
| | 0%
|
|================== | 25%
|
|=================================== | 50%
|
|==================================================== | 75%
|
|======================================================================| 100%
注意:默认情况下,在归一化、调整方差和回归掉无意义的变异源后,SCTransform将按残余方差对基因进行排序,并输出3000个变异最大的基因。如果数据集有较大的细胞数,那么使用variable.features.n参数把这个参数调高可能会有好处。
注意,最后一行输出指定了 “Set default assay to SCT”。我们可以查看我们存储在seurat对象中的不同测定。
# Check which assays are stored in objects
split_seurat$ctrl@assays
## $RNA
## Assay data with 14065 features for 14847 cells
## First 10 features:
## AL627309.1, AL669831.5, LINC00115, FAM41C, NOC2L, KLHL17, PLEKHN1,
## HES4, ISG15, AGRN
##
## $SCT
## SCTAssay data with 13799 features for 14847 cells, and 1 SCTModel(s)
## Top 10 variable features:
## FTL, CCL2, IGKC, GNLY, IGLC2, TIMP1, CCL3, IGHM, CCL4, CCL7
现在我们可以看到,除了原始RNA计数外,现在我们的assays slot中还有一个SCT组件。The most variable features是储存在SCT assays里面的唯一基因。当我们通过scRNA-seq分析时,我们将选择最合适的 assay 用于分析的不同步骤。
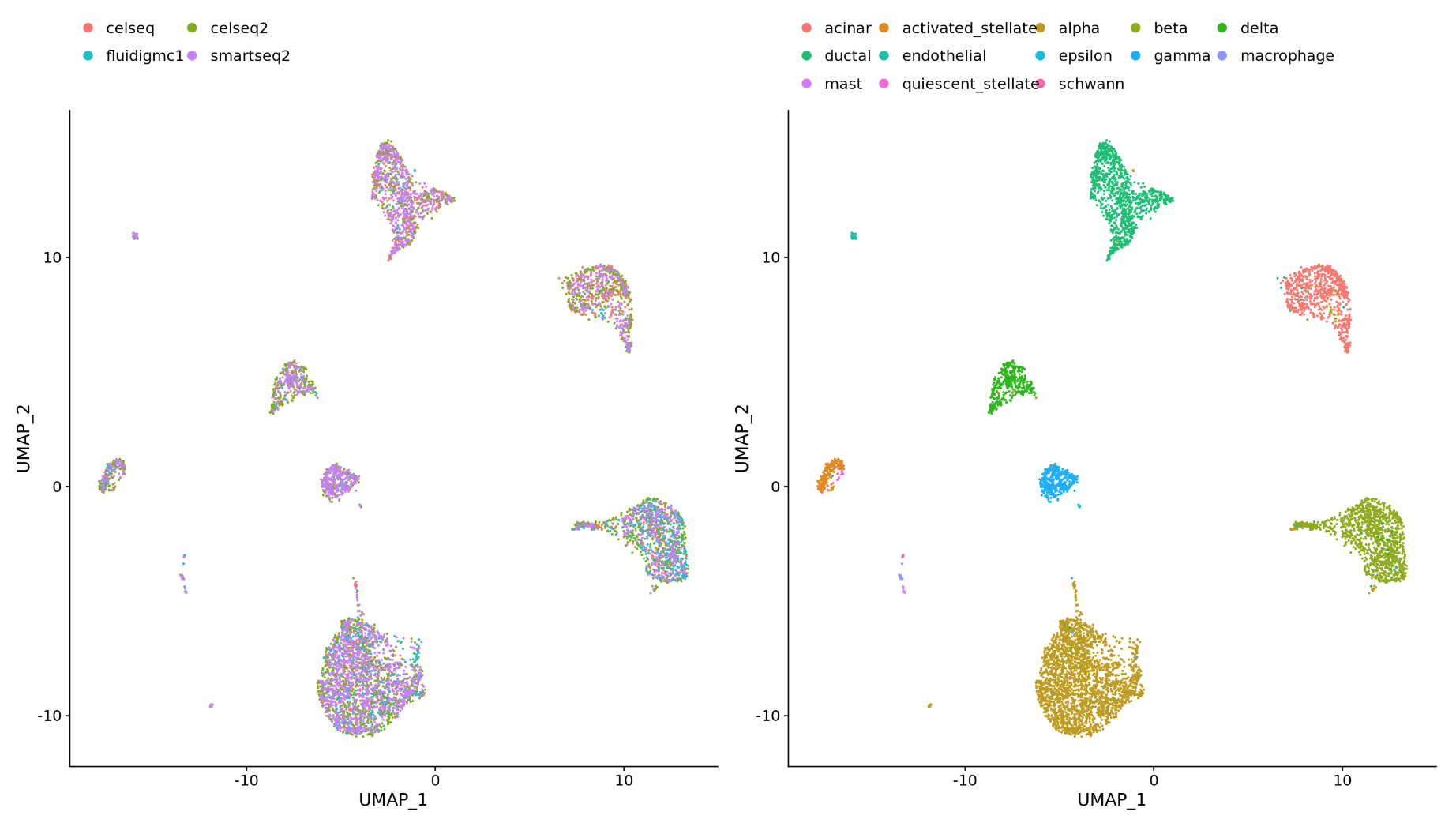
一般来说,我们总是先看我们的细胞,然后再决定是否需要进行整合。如果我们在Seurat对象中对两种conditions一起进行归一化,并将细胞之间的相似性可视化,我们就会看到特定条件的聚类。
特定条件下的细胞聚集表明,我们需要整合不同条件下的细胞。
注意:Seurat有一个关于如何在不整合的情况下运行工作流程的 vignette。该工作流程与此工作流程相当相似,但样本在开始时不一定会被分割,也不会进行整合。
2. Integrate samples using shared highly variable genes
If the cells cluster by sample, condition, dataset, or modality, this step can greatly improve your clustering and your downstream analyses.如果不确定期待什么样的聚类或期待不同条件下的一些不同的细胞类型(如肿瘤和对照样本),首先单独运行条件,然后一起运行,看是否有条件特定的聚类细胞类型出现在两个条件中,这可能会有帮助。通常情况下,当对多个条件下的细胞进行聚类时,会有特定条件下的聚类,整合可以帮助确保相同的细胞类型聚类。
为了整合,我们将使用SCTransform确定的每个条件的共享高变异基因,然后,我们将 “integrate”或 “harmonize”条件,以覆盖组之间相似或具有 “共同生物特征集”的细胞。这些组可以代表。
- 不同的条件(如控制和刺激)。

- 不同的数据集(如scRNA-seq来自同一样品上使用不同文库制备方法产生的数据集)。
- 不同的模式(如scRNA-seq和scATAC-seq)。

整合是一种强大的方法,它利用这些最大变异的共享来源来识别不同条件或数据集的共享亚群[Stuart和Bulter等人(2018)]。整合的目的是确保一个条件/数据集的细胞类型与其他条件/数据集的相同细胞类型一致(例如,对照组巨噬细胞与受刺激的巨噬细胞一致)。
具体来说,这种整合方法期望在各组中至少有一个单细胞子集的 “对应”或共享生物状态。整合分析的步骤在下图中有所概述。
Image credit: Stuart T and Butler A, et al. Comprehensive integration of single cell data, bioRxiv 2018 (https://doi.org/10.1101/460147)
应用的不同步骤如下:
- Perform canonical correlation analysis (CCA):
CCA 确定条件/组之间的共享变异源。它是 PCA 的一种形式,因为它可以识别数据中最大的变异来源,但前提是它在条件/组之间共享或保守(使用来自每个样本的 3000 个变异最大的基因)。
这一步是利用最大的共享变异源对细胞进行大致的排列。
注意:使用共享的高变异基因是因为它们最可能代表那些区分存在不同细胞类型的基因。
- Identify anchors or mutual nearest neighbors (MNNs) across datasets (sometimes incorrect anchors are identified):
MNNs可以被认为是 “best buddies”。对于一个条件下的每个细胞。
- 在另一种情况下,细胞的最近邻居是根据基因表达值确定的——它是“最好的伙伴”。
- 执行相互分析,如果两个单元格在两个方向上都是“best buddies”,那么这些单元格将被标记为锚点以将两个数据集“锚定”在一起。
“MNN 对中细胞之间表达值的差异提供了批量效应的估计,通过对许多这样的对进行平均,可以更加精确。获得校正向量并将其应用于表达值以执行批量校正。” Stuart and Bulter et al. (2018)。
- Filter anchors to remove incorrect anchors:
Assess the similarity between anchor pairs by the overlap in their local neighborhoods (incorrect anchors will have low scores) - do the adjacent cells have ‘best buddies’ that are adjacent to each other?
- Integrate the conditions/datasets:
使用锚和相应的分数来转换细胞表达值,允许条件/数据集的整合(different samples, conditions, datasets, modalities)。
注意:每个细胞的转换使用每个锚的两个细胞的加权平均值,跨数据集的锚。权重由细胞相似度得分(细胞与k个最近的锚点之间的距离)和锚点得分决定,因此同一邻域的细胞应该有相似的修正值。
如果细胞类型出现在一个数据集中,但没有出现在另一个数据集中,那么这些细胞仍将作为一个单独的特定样本集群出现。
现在,使用我们的SCTransform对象作为输入,让我们进行跨条件的整合。
首先,我们需要指定我们要使用由SCTransform确定的所有3000个最易变的基因进行整合。默认情况下,这个函数只选择前2000个基因。
# Select the most variable features to use for integration
integ_features <- SelectIntegrationFeatures(object.list = split_seurat,
nfeatures = 3000)
然后,我们需要准备SCTransform对象进行整合。
# Prepare the SCT list object for integration
split_seurat <- PrepSCTIntegration(object.list = split_seurat,
anchor.features = integ_features)
现在,我们要进行CCA,找到最好的伙伴或锚点,并过滤不正确的锚点。对于我们的数据集,这将需要长达15分钟的时间来运行。另外,请注意,你的控制台中的进度条将停留在0%,但要知道它实际上正在运行。
# Find best buddies - can take a while to run
integ_anchors <- FindIntegrationAnchors(object.list = split_seurat,
normalization.method = "SCT",
anchor.features = integ_features)
Finally, we can integrate across conditions.
# Integrate across conditions
seurat_integrated <- IntegrateData(anchorset = integ_anchors,
normalization.method = "SCT")
This would often be a good place to save the R object.
# Save integrated seurat object
saveRDS(seurat_integrated, "results/integrated_seurat.rds")
3. UMAP的可视化
在整合之后,为了使整合后的数据可视化,我们可以使用降维技术,如PCA和Uniform Manifold Approximation and Projection (UMAP)。虽然PCA会确定所有的PC,但我们一次只能绘制两个。相比之下,UMAP将从任何数量的顶级PC中获取信息,在这个多维空间中排列单元。它将把多维空间中的这些距离,试着在二维空间中绘制出来。通过这种方式,细胞之间的距离代表了表达的相似性。
为了生成这些visualizations,我们需要首先运行PCA和UMAP方法。让我们从PCA开始。
# Run PCA
seurat_integrated <- RunPCA(object = seurat_integrated)
# Plot PCA
PCAPlot(seurat_integrated,
split.by = "sample")

我们可以看到,通过PCA映射,我们对两种情况都有很好的叠加。
现在,我们也可以用UMAP进行可视化。让我们运行该方法并绘制。
# Run UMAP
seurat_integrated <- RunUMAP(seurat_integrated,
dims = 1:40,
reduction = "pca")
# Plot UMAP
DimPlot(seurat_integrated)

同样,我们看到两个条件用两种方法都能很好地对齐。有时,如果我们在不同的条件之间分割绘图,会更容易看到所有的单元格是否对齐,我们可以通过在DimPlot()函数中添加split.by参数来做到这一点。
DimPlot(seurat_integrated,
split.by = "sample")
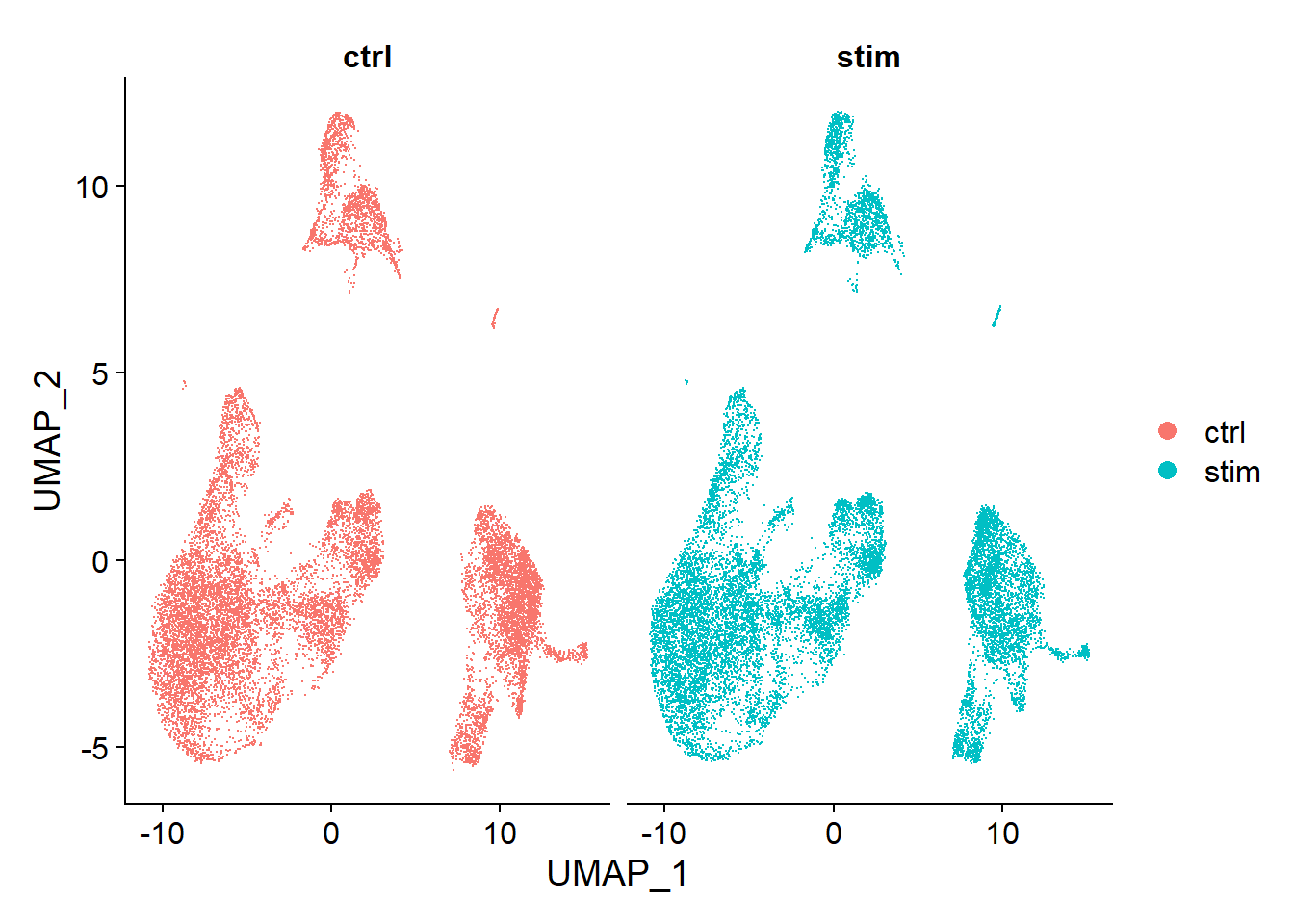
当我们与未整合的数据集进行比较时,很明显,这个数据集从整合中受益了! 
These materials have been developed by members of the teaching team at the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.

若有收获,就点个赞吧
0 人点赞

{kind=link}