TCGA下载数据,并注释
#下载数据proj = "TCGA-HNSC"download.file(url = paste0("https://gdc.xenahubs.net/download/",proj, ".htseq_counts.tsv.gz"),destfile = paste0(proj,".htseq_counts.tsv.gz"))#表达矩阵行名ID转换dat = read.table(paste0(proj,".htseq_counts.tsv.gz"),check.names = F,row.names = 1,header = T)##逆转logdat = as.matrix(2^dat - 1)dat[1:4,1:4]### 深坑一个dat[97,9]as.character(dat[97,9])### 用apply转换为整数矩阵exp = apply(dat, 2, as.integer)exp[1:4,1:4] #行名消失rownames(exp) = rownames(dat)exp[1:4,1:4]#表达矩阵过滤nrow(exp)##常用过滤标准1:###仅去除在所有样本里表达量都为零的基因exp1 = exp[rowSums(exp)>0,]nrow(exp1)##常用过滤标准2(推荐):###仅保留在一半以上样本里表达的基因exp = exp[apply(exp, 1, function(x) sum(x > 0) > 0.5*ncol(exp)), ]nrow(exp)#分组信息获取##根据样本ID的第14-15位,给样本分组(tumor和normal)library(dplyr)library(tidyverse)table(str_sub(colnames(exp),14,15))###分组Group = ifelse(as.numeric(str_sub(colnames(exp),14,15)) < 10,'tumor','normal')Group = factor(Group,levels = c("normal","tumor"))table(Group)save(exp,Group,proj,file = paste0(proj,".Rdata"))
标准化数据
rm(list = ls())load("TCGA-HNSC.Rdata")table(Group)#deseq2----library(DESeq2)colData <- data.frame(row.names =colnames(exp),condition=Group)if(!file.exists(paste0(proj,"_dd.Rdata"))){dds <- DESeqDataSetFromMatrix(countData = exp,colData = colData,design = ~ condition)dds <- DESeq(dds)save(dds,file = paste0(proj,"_dd.Rdata"))}load(file = paste0(proj,"_dd.Rdata"))class(dds)res <- results(dds, contrast = c("condition",rev(levels(Group))))c("condition",rev(levels(Group)))class(res)DEG1 <- as.data.frame(res)DEG1 <- DEG1[order(DEG1$pvalue),]DEG1 = na.omit(DEG1)head(DEG1)#添加change列标记基因上调下调logFC_t = 2pvalue_t = 0.05k1 = (DEG1$pvalue < pvalue_t)&(DEG1$log2FoldChange < -logFC_t);table(k1)k2 = (DEG1$pvalue < pvalue_t)&(DEG1$log2FoldChange > logFC_t);table(k2)DEG1$change = ifelse(k1,"DOWN",ifelse(k2,"UP","NOT"))table(DEG1$change)head(DEG1)#edgeR----library(edgeR)dge <- DGEList(counts=exp,group=Group)dge$samples$lib.size <- colSums(dge$counts)dge <- calcNormFactors(dge)design <- model.matrix(~Group)dge <- estimateGLMCommonDisp(dge, design)dge <- estimateGLMTrendedDisp(dge, design)dge <- estimateGLMTagwiseDisp(dge, design)fit <- glmFit(dge, design)fit <- glmLRT(fit)DEG2=topTags(fit, n=Inf)class(DEG2)DEG2=as.data.frame(DEG2)head(DEG2)k1 = (DEG2$PValue < pvalue_t)&(DEG2$logFC < -logFC_t);table(k1)k2 = (DEG2$PValue < pvalue_t)&(DEG2$logFC > logFC_t);table(k2)DEG2$change = ifelse(k1,"DOWN",ifelse(k2,"UP","NOT"))head(DEG2)table(DEG2$change)###limma----library(limma)dge <- DGEList(counts=exp)dge <- calcNormFactors(dge)v <- voom(dge,design, normalize="quantile")design <- model.matrix(~Group)fit <- lmFit(v, design)fit= eBayes(fit)DEG3 = topTable(fit, coef=2, n=Inf)DEG3 = na.omit(DEG3)k1 = (DEG3$P.Value < pvalue_t)&(DEG3$logFC < -logFC_t);table(k1)k2 = (DEG3$P.Value < pvalue_t)&(DEG3$logFC > logFC_t);table(k2)DEG3$change = ifelse(k1,"DOWN",ifelse(k2,"UP","NOT"))table(DEG3$change)head(DEG3)tj = data.frame(deseq2 = as.integer(table(DEG1$change)),edgeR = as.integer(table(DEG2$change)),limma_voom = as.integer(table(DEG3$change)),row.names = c("down","not","up"));tjsave(DEG1,DEG2,DEG3,Group,tj,file = paste0(proj,"_DEG.Rdata"))
PCA图
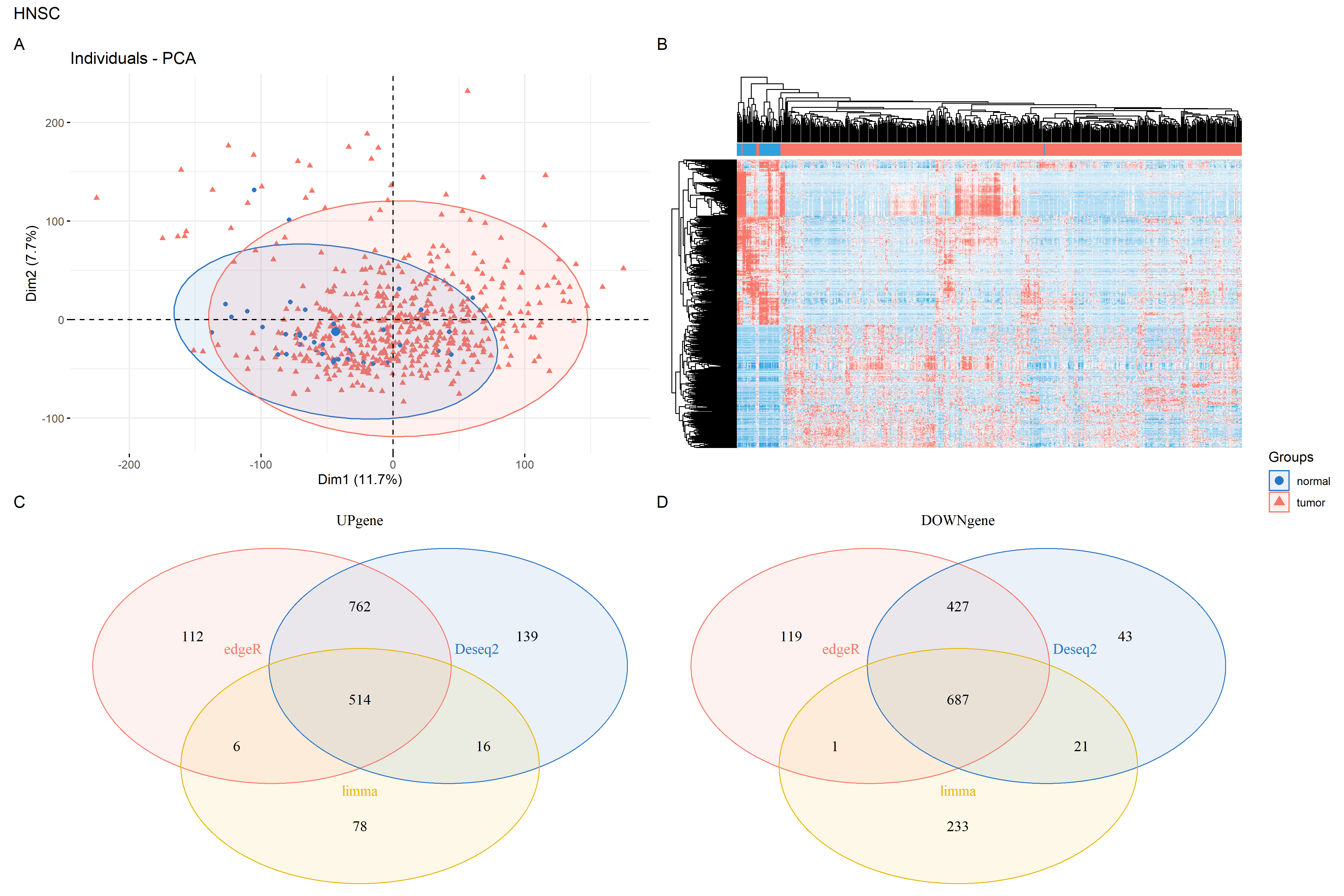
rm(list = ls())library(ggplot2)library(tinyarray)load("TCGA-HNSC.Rdata")load("TCGA-HNSC_DEG.Rdata")exp[1:4,1:4]ncol(exp)# cpm 去除文库大小的影响dat = log2(cpm(exp)+1)pca.plot = draw_pca(dat,Group);pca.plotsave(pca.plot,file = paste0(proj,"_pcaplot.Rdata"))
TCGA作图,拼图
install.packages('VennDiagram')library('VennDiagram')UP=function(df){rownames(df)[df$change=="UP"]}DOWN=function(df){rownames(df)[df$change=="DOWN"]}up = intersect(intersect(UP(DEG1),UP(DEG2)),UP(DEG3))down = intersect(intersect(DOWN(DEG1),DOWN(DEG2)),DOWN(DEG3))dat = log2(cpm(exp)+1)hp = draw_heatmap(dat[c(up,down),],Group,n_cutoff = 2)#上调、下调基因分别画维恩图up_genes = list(Deseq2 = UP(DEG1),edgeR = UP(DEG2),limma = UP(DEG3))down_genes = list(Deseq2 = DOWN(DEG1),edgeR = DOWN(DEG2),limma = DOWN(DEG3))up.plot <- draw_venn(up_genes,"UPgene")down.plot <- draw_venn(down_genes,"DOWNgene")#维恩图拼图,终于搞定library(patchwork)#up.plot + down.plot# 拼图pca.plot + hp+up.plot +down.plot+ plot_layout(guides = "collect")+plot_annotation(title = "HNSC",tag_levels = "A")ggsave(paste0(proj,"_heat_ve_pca.png"),width = 15,height = 10)

若有收获,就点个赞吧
0 人点赞
