摘要:病理图像在患者预后、肿瘤分类分级等医学问题中扮演着重要角色,随着计算机技术的发展,数字病理图像分析备受关注。SVS格式是常见的储存病理图像的方式,本文介绍了使用Python作为工具,对SVS格式病理图像进行预处理的方式。
一、WSI(Whole-Slide Image)读取
“工欲善其事,必先利其器”,首先需要安装一个常用的处理病理图像的Python库。
全扫描WSI病理图像文件特别大,OpenSlide提供了处理这类SVS文件的接口,使用pip安装命令如下:
pip install openslide-python
读取方式为:
import openslideinput_img_path = ''original = openslide.OpenSlide(input_img_path)
将HistomicsTk安装为Python的一个工具包,linux系统中安装过程如下:
pip install histomicstk --find-links https://girder.github.io/large_image_wheels
该工具包提供了分析病理图像的接口,是基于Digital Slide Archive和large_image构建的,其中large_iamge可用于SVS文件的读取。
import large_imagewsi_path = ''ts = large_image.getTileSource(wsi_path)print(ts.getMetadata())print(ts.getNativeMagnification())# Get the magnification associated with all levels of the image pyramidfor i in range(ts.levels):print('Level-{} : {}'.format(i, ts.getMagnificationForLevel(level=i)))
举例结果如下:
{'magnification': 20.0, 'levels': 8, 'tileHeight': 256, 'tileWidth': 256, 'sizeX': 16000, 'sizeY': 22465, 'mm_y': 0.0005015, 'mm_x': 0.0005015}{'magnification': 20.0, 'mm_y': 0.0005015, 'mm_x': 0.0005015}Level-0 : {'magnification': 0.15625, 'scale': 128.0, 'level': 0, 'mm_y': 0.064192, 'mm_x': 0.064192}Level-1 : {'magnification': 0.3125, 'scale': 64.0, 'level': 1, 'mm_y': 0.032096, 'mm_x': 0.032096}Level-2 : {'magnification': 0.625, 'scale': 32.0, 'level': 2, 'mm_y': 0.016048, 'mm_x': 0.016048}Level-3 : {'magnification': 1.25, 'scale': 16.0, 'level': 3, 'mm_y': 0.008024, 'mm_x': 0.008024}Level-4 : {'magnification': 2.5, 'scale': 8.0, 'level': 4, 'mm_y': 0.004012, 'mm_x': 0.004012}Level-5 : {'magnification': 5.0, 'scale': 4.0, 'level': 5, 'mm_y': 0.002006, 'mm_x': 0.002006}Level-6 : {'magnification': 10.0, 'scale': 2.0, 'level': 6, 'mm_y': 0.001003, 'mm_x': 0.001003}Level-7 : {'magnification': 20.0, 'level': 7, 'scale': 1.0, 'mm_y': 0.0005015, 'mm_x': 0.0005015}
这反应了WSI病理图像的层级结构,如图一所示,不同的Level代表着不同的分辨率,对于此例而言,最高放大倍数为20倍。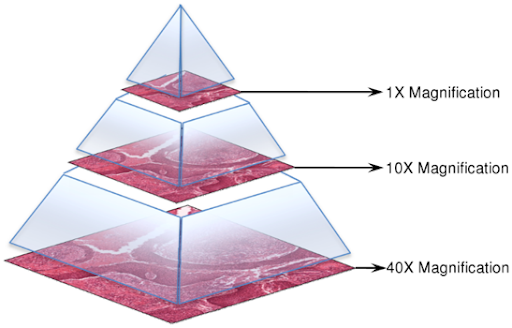
图一、病理图像的金字塔层级结构
二、WSI中patchs切片
直接对高分辨度的WSI进行分析处理是不可行的,因此通常需要将整张全扫描图像分割成patches,然后对感兴趣部位的ROI区域进行分析。以下展示了如何从全扫描WSI中选取密度最高的10张20倍放大倍数patches,每张patch尺寸为1024*1024,并保存。
import large_image
import os
import heapq
import numpy as np
import pandas as pd
from tqdm import tqdm, trange
import matplotlib.pyplot as plt
from PIL import Image
def calc_density(image_chunk):
pixels = np.array(image_chunk)
return float(np.sum(pixels < 200)) / pixels.size
csv_path = 'LUAD_img_adress.csv' #该文件保存了所有病理图像svs文件地址
csv_data = pd.read_csv(csv_path)
csv_df = pd.DataFrame(csv_data)
img_preprocess = './LUAD_img_preprocess'
num_img = 0
for index, row in csv_df.iterrows():
out_dir = os.path.join(img_preprocess,row["patient_id"]+"_large_image")
if not os.path.exists(out_dir):
os.mkdir(out_dir)
wsi_path = row["image_adress"]
ts = large_image.getTileSource(wsi_path)
top10 = []
num_tile = 0
for tile_info in ts.tileIterator(
scale=dict(magnification=20),
tile_size=dict(width=1024, height=1024),
tile_overlap=dict(x=50, y=50),
format=large_image.tilesource.TILE_FORMAT_PIL
):
im_array = np.array(tile_info['tile'])
if im_array.shape == (1024,1024,4):
density_score = calc_density(tile_info['tile'])
entry = (density_score, num_tile, tile_info['tile'])
if len(top10) < 10:
heapq.heappush(top10, entry)
elif density_score > top10[0][0]:
heapq.heappushpop(top10, entry)
num_tile += 1
for j, entry in enumerate(top10):
im_tile = Image.fromarray(np.array(entry[2]))
im_tile.save(os.path.join(out_dir,str(j) + '.png'))
三、Hematoxylin和Eosin染色分解
常见的病理图像是由H&E方法进行染色的,其中Hematoxylin主要对遗传物质DNA具有着色作用,而Eosin主要是对蛋白进行着色,将这病理图像中的这两种染色进行分解,有利于后续进一步分析。
import matplotlib
matplotlib.use('Agg')
import matplotlib.pyplot as plt
import matplotlib.patches as mpatches
from PIL import Image
import openslide
import sys
import os
import logging
import numpy as np
import pandas as pd
from tqdm import trange
import concurrent.futures
import histomicstk as htk
import numpy as np
import scipy as sp
import skimage.io
import skimage.measure
import skimage.color
titlesize = 24
input_image_file = 'patch-demo.png'
im_input = skimage.io.imread(input_image_file)[:, :, :3]
# create stain to color map
stainColorMap = {
'hematoxylin': [0.65, 0.70, 0.29],
'eosin': [0.07, 0.99, 0.11],
'dab': [0.27, 0.57, 0.78],
'null': [0.0, 0.0, 0.0]
}
# specify stains of input image
stain_1 = 'hematoxylin' # nuclei stain
stain_2 = 'eosin' # cytoplasm stain
stain_3 = 'null' # set to null of input contains only two stains
# create stain matrix
W = np.array([stainColorMap[stain_1],
stainColorMap[stain_2],
stainColorMap[stain_3]]).T
# perform standard color deconvolution
im_stains = htk.preprocessing.color_deconvolution.color_deconvolution(im_input, W).Stains
# Display results
plt.figure(figsize=(20, 10))
plt.subplot(1, 2, 1)
plt.imshow(im_stains[:, :, 0])
plt.title(stain_1, fontsize=titlesize)
plt.subplot(1, 2, 2)
plt.imshow(im_stains[:, :, 1])
_ = plt.title(stain_2, fontsize=titlesize)
plt.savefig('HEdecolor.png')
按两种染色剂将图像分解结果如图三所示:
图三、Hematoxylin和Eosin染色分解结果示例
四、Nuclei识别及形态学特征提取
病理图像中Nuclei的识别以及相应形态学特征对于疾病的预后起到很重要的辅助作用。以下过程将基于第三步中分解出的Hematoxylin通道结果(Hematoxylin着重对遗传物质DNA进行染色),对图像中的Nuclei进行分析。
import matplotlib
matplotlib.use('Agg')
import matplotlib.pyplot as plt
import matplotlib.patches as mpatches
from PIL import Image
import openslide
import sys
import os
import logging
import numpy as np
import pandas as pd
from tqdm import trange
import concurrent.futures
import histomicstk as htk
import numpy as np
import scipy as sp
import skimage.io
import skimage.measure
import skimage.color
titlesize = 24
input_image_file = 'patch-demo.png’
im_input = skimage.io.imread(input_image_file)[:, :, :3]
# create stain to color map
stainColorMap = {
'hematoxylin': [0.65, 0.70, 0.29],
'eosin': [0.07, 0.99, 0.11],
'dab': [0.27, 0.57, 0.78],
'null': [0.0, 0.0, 0.0]
}
# specify stains of input image
stain_1 = 'hematoxylin' # nuclei stain
stain_2 = 'eosin' # cytoplasm stain
stain_3 = 'null' # set to null of input contains only two stains
# create stain matrix
W = np.array([stainColorMap[stain_1],
stainColorMap[stain_2],
stainColorMap[stain_3]]).T
# perform standard color deconvolution
im_stains = htk.preprocessing.color_deconvolution.color_deconvolution(im_input, W).Stains
# get nuclei/hematoxylin channel
im_nuclei_stain = im_stains[:, :, 0]
# segment foreground
foreground_threshold = 60
im_fgnd_mask = sp.ndimage.morphology.binary_fill_holes(
im_nuclei_stain < foreground_threshold)
# run adaptive multi-scale LoG filter
min_radius = 5
max_radius = 15
im_log_max, im_sigma_max = htk.filters.shape.cdog(
im_nuclei_stain, im_fgnd_mask,
sigma_min=min_radius * np.sqrt(2),
sigma_max=max_radius * np.sqrt(2)
)
# detect and segment nuclei using local maximum clustering
local_max_search_radius = 10
im_nuclei_seg_mask, seeds, maxima = htk.segmentation.nuclear.max_clustering(
im_log_max, im_fgnd_mask, local_max_search_radius)
# filter out small objects
min_nucleus_area = 5
im_nuclei_seg_mask = htk.segmentation.label.area_open(
im_nuclei_seg_mask, min_nucleus_area).astype(np.int)
# compute nuclei properties
#objProps = skimage.measure.regionprops(im_nuclei_seg_mask)
objProps = skimage.measure.regionprops(im_nuclei_seg_mask)
print 'Number of nuclei = ', len(objProps)
# Display results
plt.figure(figsize=(20, 10))
plt.subplot(1, 2, 1)
plt.imshow(skimage.color.label2rgb(im_nuclei_seg_mask, im_input, bg_label=0), origin='lower')
plt.title('Nuclei segmentation mask overlay', fontsize=titlesize)
plt.subplot(1, 2, 2)
plt.imshow( im_input )
plt.xlim([0, im_input.shape[1]])
plt.ylim([0, im_input.shape[0]])
plt.title('Nuclei bounding boxes', fontsize=titlesize)
for i in range(len(objProps)):
c = [objProps[i].centroid[1], objProps[i].centroid[0], 0]
width = objProps[i].bbox[3] - objProps[i].bbox[1] + 1
height = objProps[i].bbox[2] - objProps[i].bbox[0] + 1
cur_bbox = {
"type": "rectangle",
"center": c,
"width": width,
"height": height,
}
plt.plot(c[0], c[1], 'g+')
mrect = mpatches.Rectangle([c[0] - 0.5 * width, c[1] - 0.5 * height] ,
width, height, fill=False, ec='g', linewidth=2)
plt.gca().add_patch(mrect)
plt.savefig('getNuclei-demo.png')
a = htk.features.compute_morphometry_features(im_nuclei_seg_mask)
d = a.mean().to_dict()
morphometry_df = pd.DataFrame(d,index=[0])
morphometry_df.to_csv('morphometry-features-demo.csv')
Number of nuclei = 1115
本张patch中共识别到1115个nuclei,识别结果如图四所示:
图四、Nuclei识别结果
提取到的形态学特征如表一
| Shape.Circularity | Shape.Eccentricity | Shape.EquivalentDiameter | Shape.Extent | Shape.MinorMajorAxisRatio | Shape.Solidity | Size.Area | Size.MajorAxisLength | Size.MinorAxisLength | Size.Perimeter |
|---|---|---|---|---|---|---|---|---|---|
| 0.5609776331378570 | 0.7984962420761610 | 14.587835871173700 | 0.37756252203272500 | 0.5614023145516780 | 0.5707893698856090 | 201.1291479820630 | 25.850580121724300 | 14.221723122921500 | 84.91634679492940 |
表一、Morphometry features
未来可以基于这些提取到的特征进行组织分类以及后续生存分析。

若有收获,就点个赞吧
0 人点赞
