预计目标
- 让不了解Mega的使用者看完文章之后可以进行简单的操作。
- 完成DNA的对比。
开始
- 下载Mega7软件,建议7.0.26版本(可看参考【2】)。
启动软件如下图:
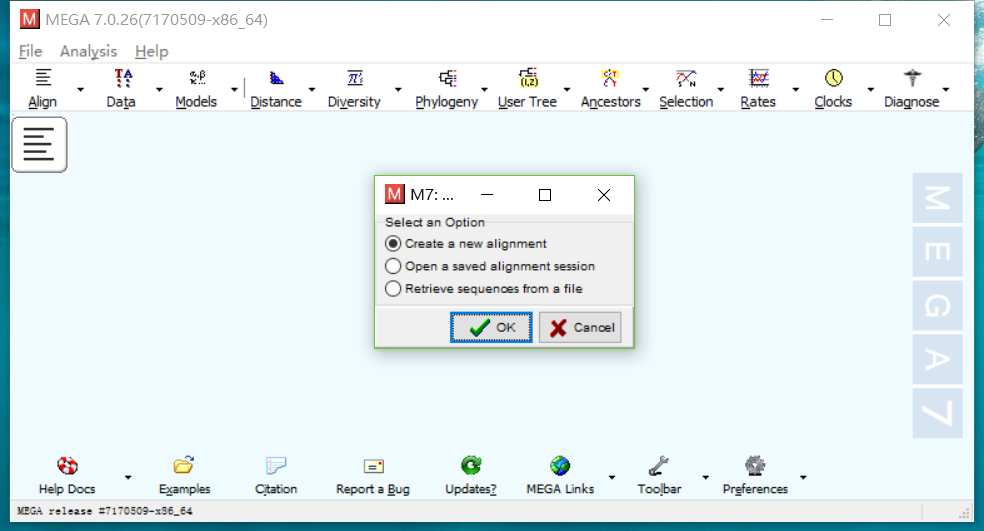
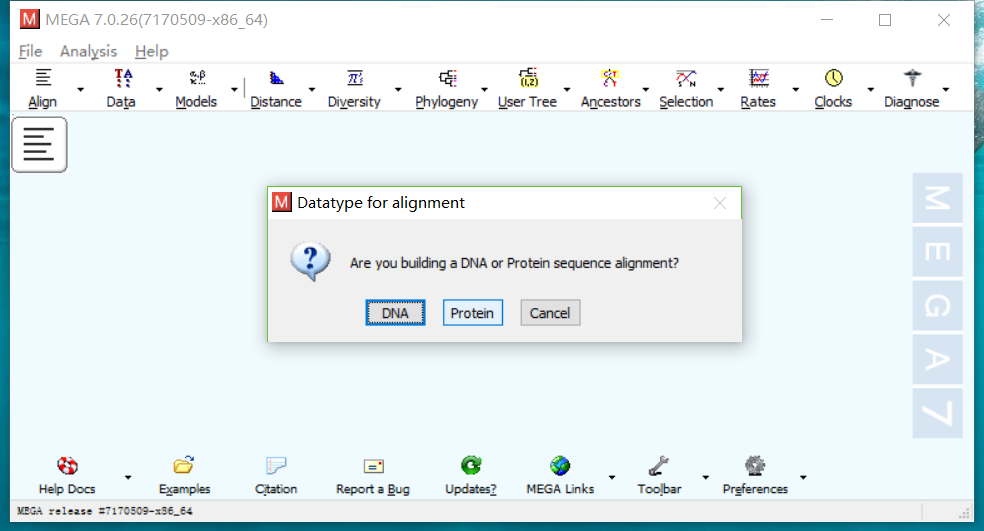
- 点击Align->Edit/Build Alignment,新建一个对比,选择DNA。
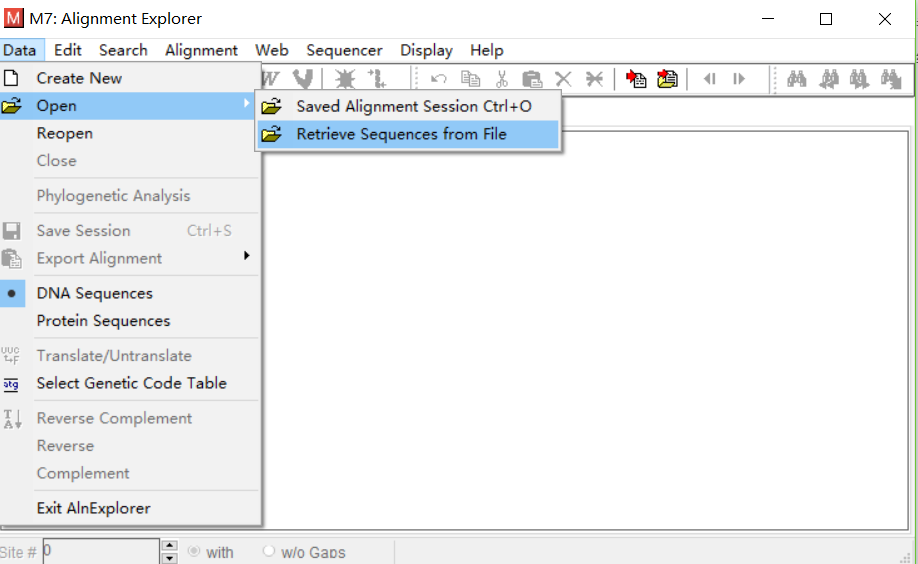
- 点击Data->Open->Retrieve Sequences from File
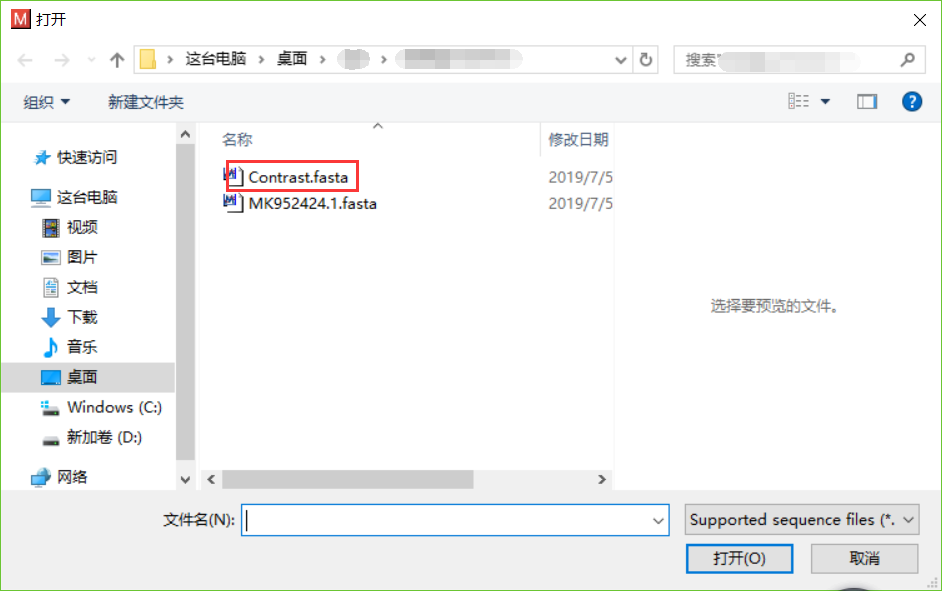
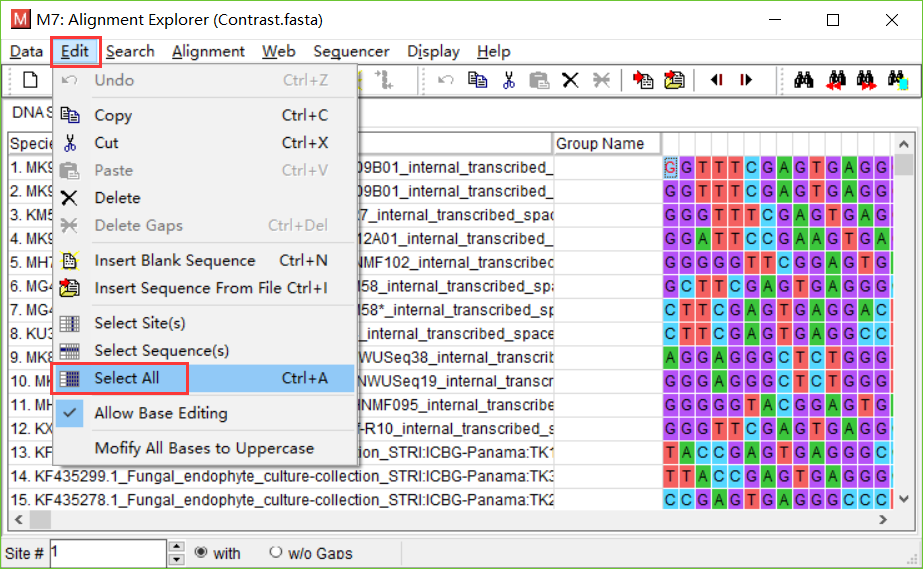
- 打开需要处理的fasta文件,点击Edit->Select All,点击Alignment->Align by ClustalW方法。

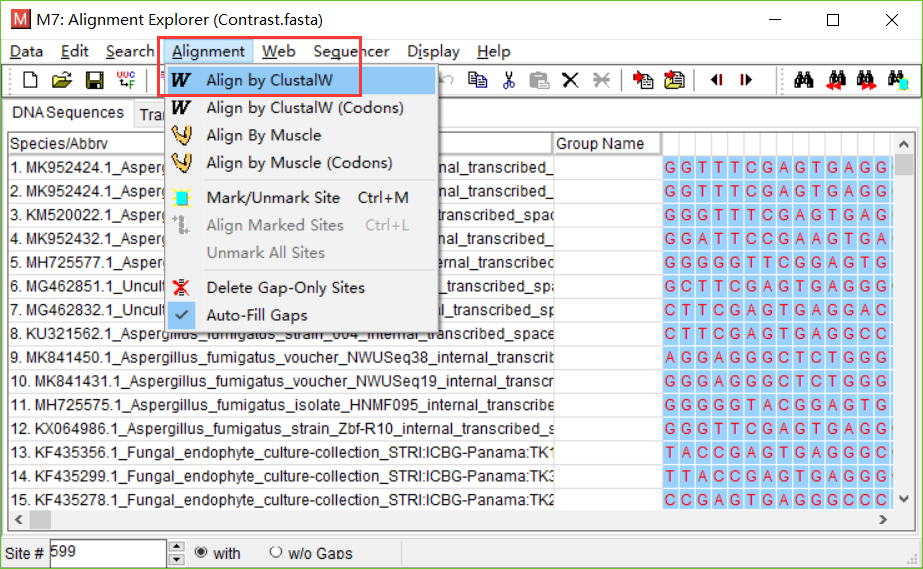
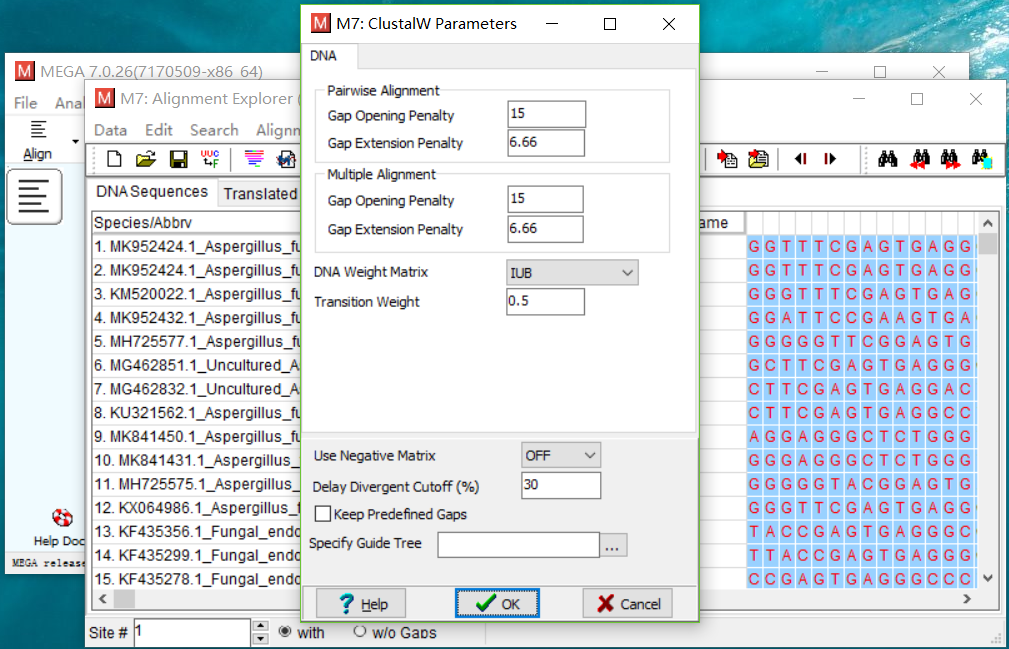
点击确定,参数使用默认的。
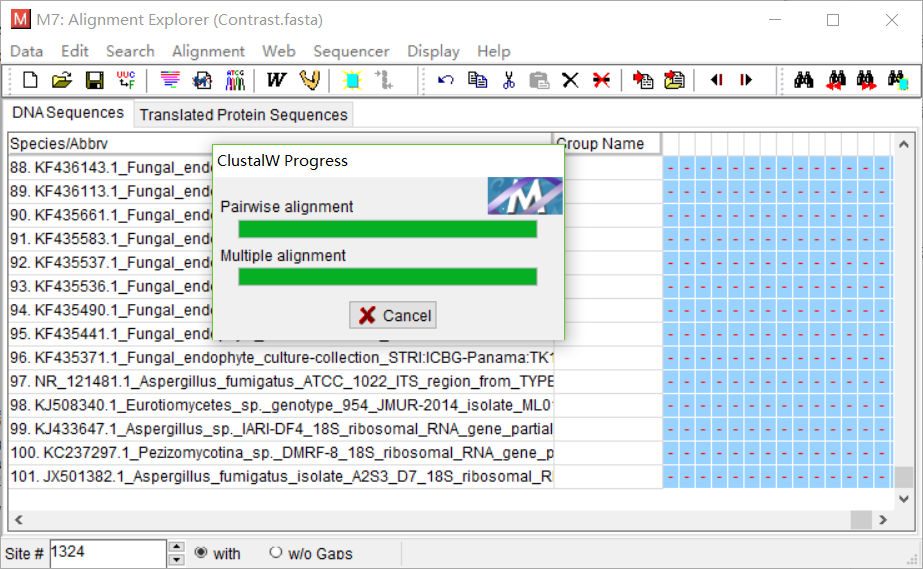
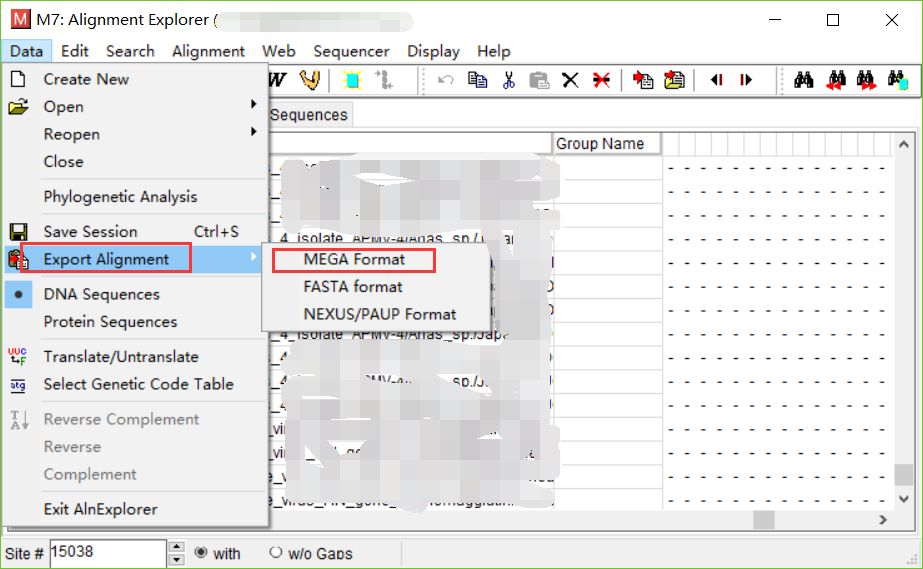
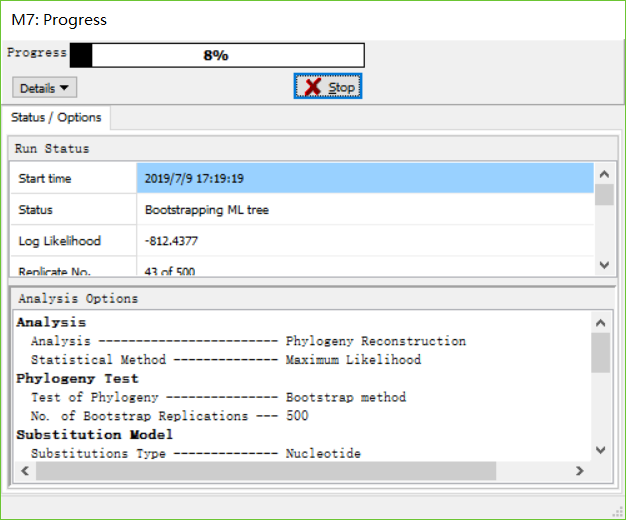
当数据跑完之后,保存数据,保存为meg格式。
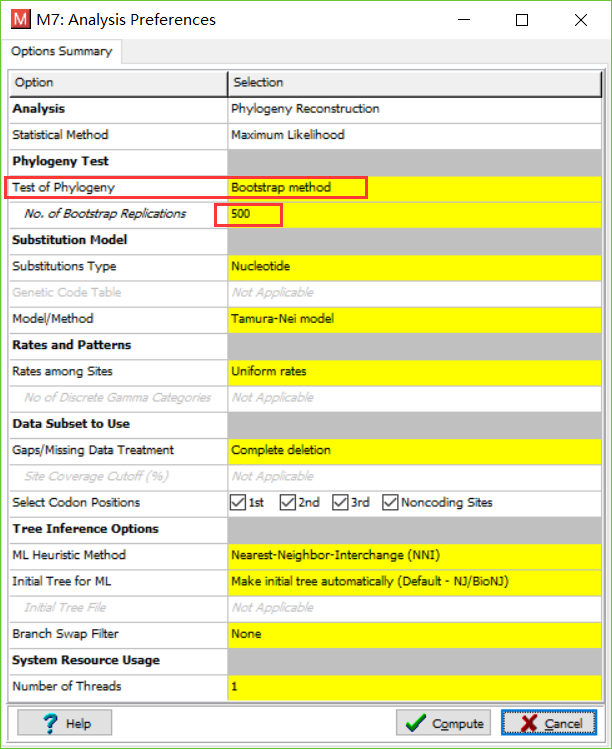
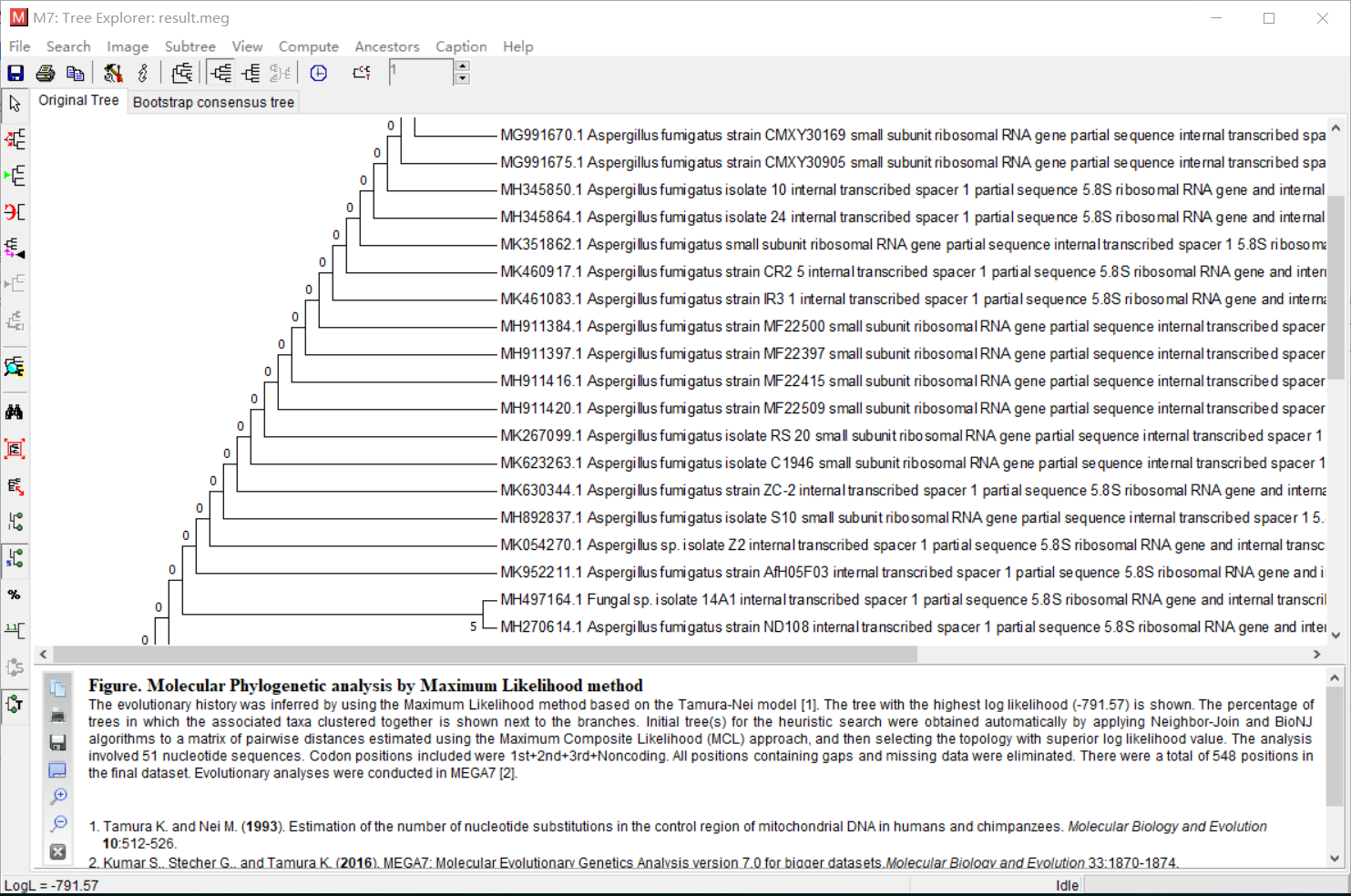
- 展示数据。将刚刚保存的数据打开,并且完成绘图。作者使用的是ML法。

- 之后会得到对比的结果图。
获取对比的序列
将自己的序列添加进去,点击BLAST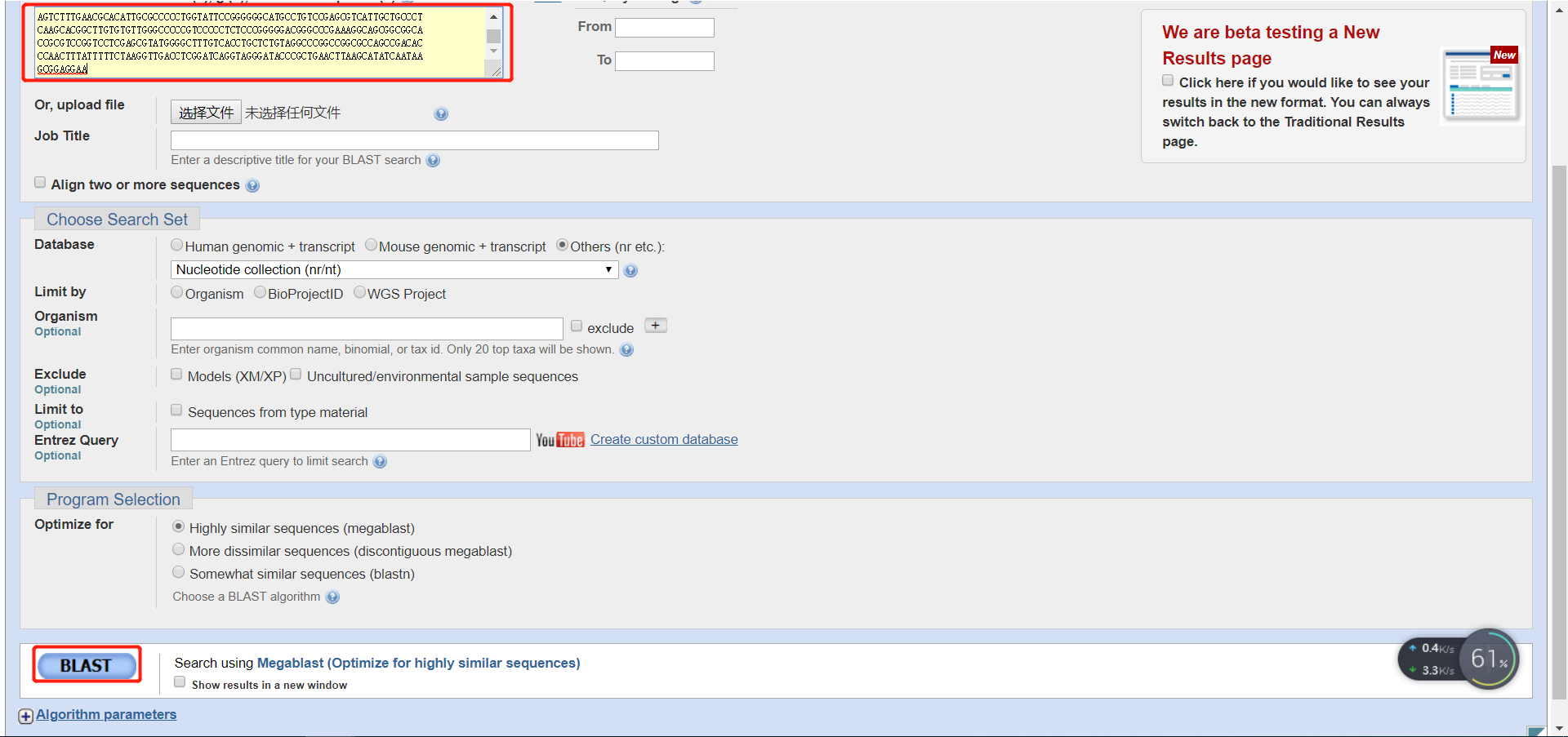
保存结果为fasta格式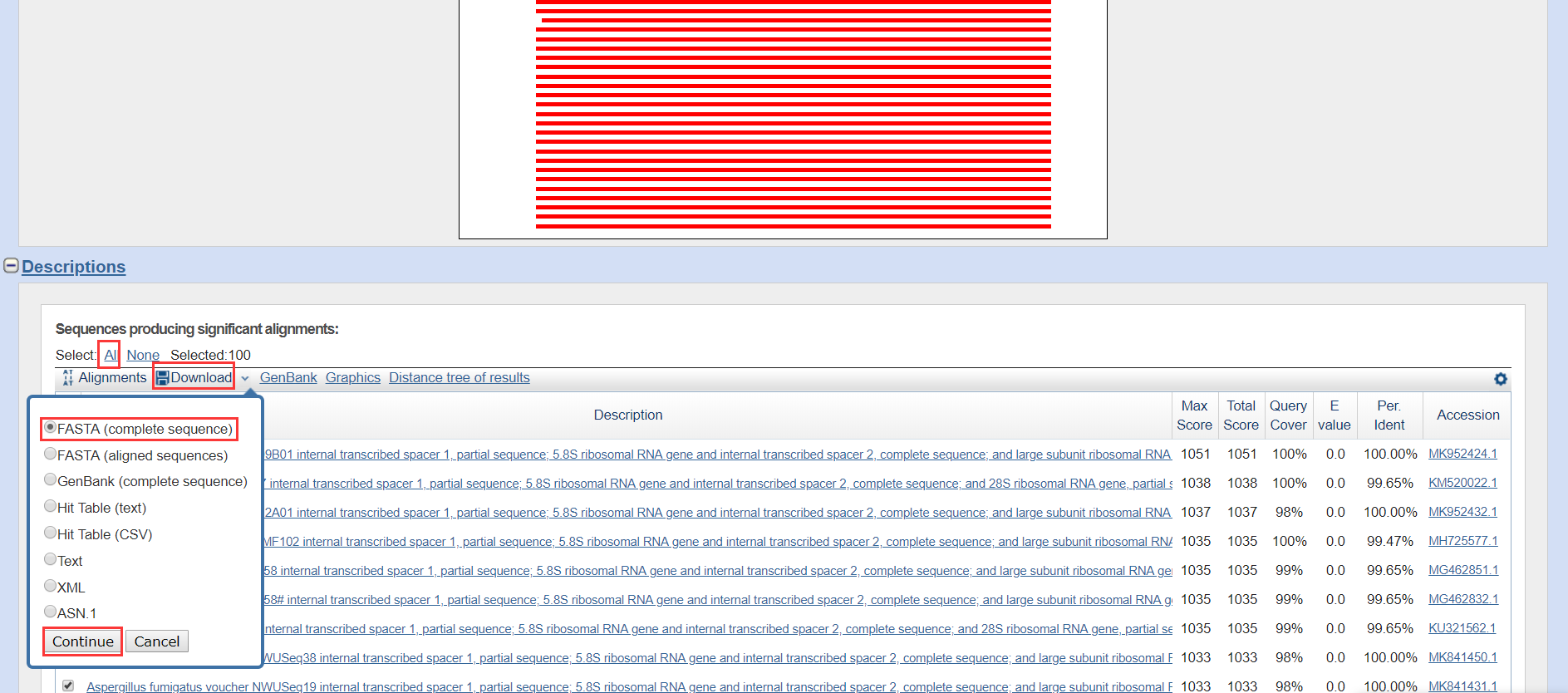
比较方法
构建系统进化树的理论方法主要有基于距离的方法UPGMA、ME(Minimum Evolution,最小进化法)和NJ(Neighbor-Joining,邻接法)等。此外,还包括MP(Maximum parsimony,最大简约法)、ML(Maximum likelihood,最大似然法)以及贝叶斯(Bayesian)推断等方法。其中UPGMA法已经较少使用。下面对其中最常用的3种方法简要介绍。
1、邻接法(Neighbor-Joining,NJ):
该方法通过确定距离最近或相邻的成对分类单位来使系统树的总距离达到最小。相邻是指两个分类单位在某一无根分叉树中仅通过一个节点(Node)相连。通过循序地将相邻点合并成新的点,就可以建立一个相应的拓扑树。
2、最大似然法(Maximum likelihood,ML):
最早应用于系统发育分析是在对基因频率数据的分析上,后来基于分子序列的分析中也引入了最大似然法的分析方法。最大似然法分析中,选取一个特定的替代模型来分析给定的一组序列数据,使得获得的每一个拓扑结构的似然率都为最大值,然后再挑出其中似然率最大的拓扑结构作为最优树。在最大似然法的分析中,所考虑的参数并不是拓扑结构而是每个拓扑结构的枝长,并对似然率球最大值来估计枝长。最大似然法的建树过程是个很费时的过程,因为在分析过程中有很大的计算量,每个步骤都要考虑内部节点的所有可能性。最大似然法也是一个比较成熟的参数估计的统计学方法,具有很好的统计学理论基础,在当样本量很大的时候,似然法可以获得参数统计的最小方差。只要使用了一个合理的、正确的替代模型,最大似然法可以推导出一个很好的进化树结果。
3、最大简约法(Maximum parsimony,MP):
最早源于形态性状研究,现在已推广到分子序列的进化分析中。最大简约法的理论基础是奥卡姆(Ockham)哲学原则,这个原则认为:解释一个过程的最好理论是所需假设数目最少的那一个。对所有可能的拓扑结构进行计算,并计算出所需替代数最小的那个拓扑结构,作为最优树。优点:最大简约法对于分析某些特殊的分子数据,如插入、缺失等序列有用。在分析的序列位点上没有回复突变或平行突变,且被检验的序列位点数很大的时候,最大简约法能够推导获得一个很好的进化树。缺点:在分析序列上存在较多的回复突变或平行突变,而被检验的序列位点数又比较少的时候,最大简约法可能会给出一个不合理的或者错误的进化树推导结果。
方法总结
一般来讲,如果模型合适,ML的效果较好。对近缘序列,有人喜欢MP,因为用的假设最少。MP一般不用在远缘序列上,这时一般用NJ或ML。对相似度很低的序列,NJ往往出现Long-branch attraction(LBA,长枝吸引现象),有时严重干扰进化树的构建。贝叶斯的方法则太慢。对于各种方法构建分子进化树的准确性,一篇综述(Hall BG. Mol Biol Evol 2005, 22(3):792-802)认为贝叶斯的方法最好,其次是ML,然后是MP。其实如果序列的相似性较高,各种方法都会得到不错的结果,模型间的差别也不大。
对于NJ和ML,是需要选择模型的。对于蛋白质序列以及DNA序列,两者模型的选择是不同的。对于蛋白质的序列,一般选择Poisson Correction(泊松修正)这一模型。而对于核酸序列,一般选择Kimura 2-parameter(Kimura-2参数)模型。如果对各种模型的理解并不深入,不推荐初学者使用其他复杂的模型。
关于fasta文件格式
第一种方法
找到你的ab1格式的文件,使用 SnapGene Viewer 打开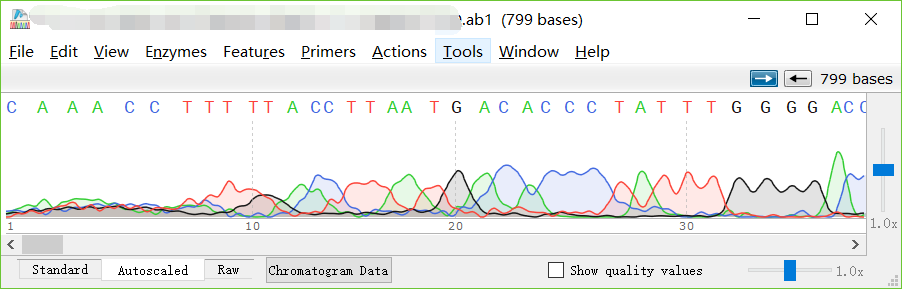
点击File->Save As->选择格式为FASTA,就保持好了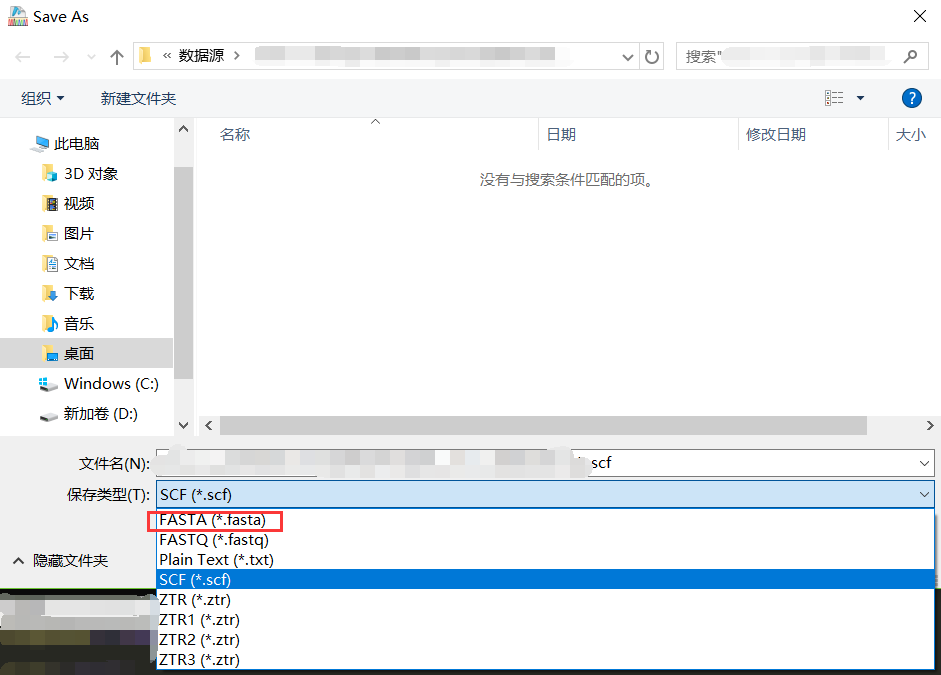
第二种方法
把存好数据的txt文件直接改成fasta就好
比如:seqdump (1).txt 改成 seqdump (1).fasta 保存
参考
- Mega软件教学
- Mege7下载地址
- 你可能还需要这个软件SnapGene Viewer
- 你可能还需要这个软件Notepad++
总结
- 电脑配置不行的话,跑NJ法容易卡死。
- 娶老婆不容易啊,还要学生物。

若有收获,就点个赞吧
0 人点赞
