会议纪要
1.根据搜索引擎retrieval的方式,去修改loss function,以期望在最后的鉴定任务中,会获得更多的鉴定肽段
2.对于同一个母体sequence,出现不同的修饰方式,从而谱图会有区别,我们去准确预测这种区别,是很有生物学意义的。
3.CNN model 的 kernel size是2,一般是奇数。
4.用Prosit 的数据去pre train 我们的CNN model和Transformer Model。对于CNN,期望我们可以通过在大的数据集上的训练,避免在Jeff小数据量上的过拟合现象。对于Transformer,rongjie师兄建议transformer需要好的预训练,我们现在采取的预训练的方式就是在prosit数据上训练。目的是为了在Jeff数据上(target 数据)上提升预测效果,而不是一定要达到在Prosit上报道的结果。
5.因为Prosit没有针对磷酸化修饰,而pdeep2是可以进行磷酸化序列的预测,所以我们的baseline是pdeep2。
6.对于我们选择的额外的feature,除了0以外,剩下的都在700以上,最高是1000,所以normalize后,是0,0.7+这样的distribution,何老师建议把700以上的值都减去700,这样拉近和0的距离(最小是775)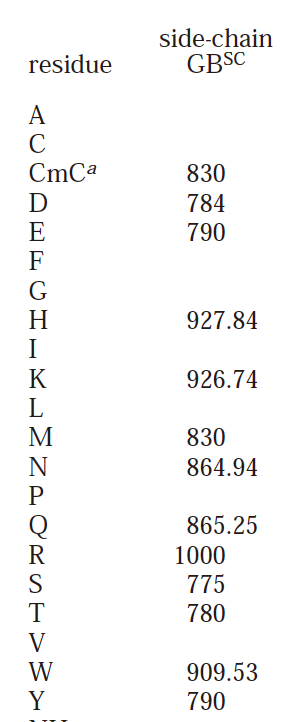
7.Model ensemble一般是将最后的结果输出做average,一般不会把hidden state concat起来。
ToDo
1.继续做上次讨论的two-stage的做法,要观察使用two-stage 前后,是否model对于0与非0的预测更加准确了。
2.对于同一个母体sequence,但是不同的修饰状态,它们相对应的谱图也是不同的,这种数据比较少,我们需要先在Jeff数据里统计下这样的数据的情况,继而去设计model和loss function
3.总的milestone分为两步,第一步是先继续延续之前的思路,在总的谱图预测上先优化PCC和SA;第二阶段去专注于同一个母体sequence,尝试去更准确预测这种fine-grained差异。
4.针对3中第一步,主要关注PCC和SA以及最终鉴定出来的肽段的值,第二步是尝试设计新的,metric去衡量我们对于fine-grained的谱图的差异性的判断,以及观察是否某种特定种类的肽段鉴定的数目增多且丢掉的变少。

若有收获,就点个赞吧
0 人点赞
