【考前提醒】:
1.导入数据后先对数据进行简单的观察,行列数,数据类型,每行每列含义,在进行计算
2.不要只会埋头敲代码,要明白每一步是在做什么,有结果后,要学会对其进行简单的检查,看命令有没有错误
3.报错时,要冷静分析错误的原因,不要一味重复进行,没有用的
2021年生统作业
第一次作业
1.计算平均数
2.计算概率
3.P(A+B)=P(A)+P(B)-P(AB)
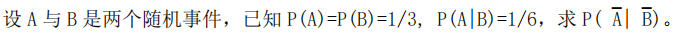
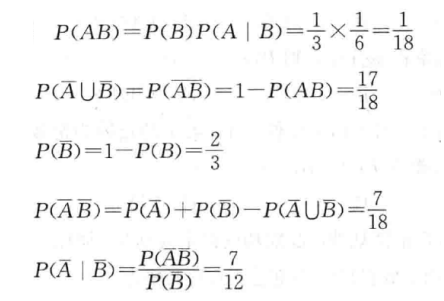
4.P(AB)=P(A)P(B|A)=P(B)P(A|B)
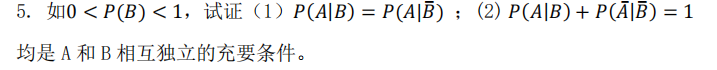
5.中心极限定理

第二次作业
1.z检验
2.概率计算
3.比例检验
4.泊松逼近
5.概率计算
6.标准正态化
第三次作业
1.R 的基本操作
#查看当前工作目录;更改工作目录到桌面> getwd()> setwd('C:/Users/cuihy/Desktop/')#下载安装 R 包> install.packages('datasets')> library(datasets)#载入 iris 数据集,并查看数据行列数,查看数据类型> data("iris")> nrow(iris)> ncol(iris)> mode(iris) 或 class(iris) 或 str(iris)#对 iris 的 Sepal.Length 进行描述统计。并绘制箱线图。> summary(iris$sepal_length)> boxplot(iris$sepal_length)#用 R 查看 t.test()的帮助文档> ?t.test
2.t.检验
#利用上一题的鸢尾花数据集进行计算。鸢尾花的平均花瓣长度为 3.5cm,这种说法正确吗?> t.test(iris$Sepal.Length,alternative = "two.sided",mu = 3.5)One Sample t-testdata: iris$Sepal.Lengtht = 34.659, df = 149, p-value < 2.2e-16alternative hypothesis: true mean is not equal to 3.595 percent confidence interval:5.709732 5.976934sample estimates:mean of x5.843333
3.文件基本操作
#查看该目录下的文件> dir()#将基因表达数据 homework3.1_data.txt 导入到 R 中,使行名为样本编号,列名为 symbol 基因名,并查看行列数及前 5 行数据, 计算每个基因表达的均值,标准差和最大值。> data3 <- read.table("D:/郭东灵/我的研究生/生统作业/homework3.1_data.txt",header=TRUE,row.names=1)> data3 <- t(data3)> dim(data)14 199> head(data, n=5)> statistic <- apply(data3,2,function(x){+ Mean <- mean(x);names(Mean)<-"Mean"+ Sd<-sd(x);names(Sd)<-"Sd"+ Max<-max(x);names(Max)<-"Max"+ return(c(Mean,Sd,Max))+ }) ;apply函数行、列运算#对样本 A9QA 的基因表达进行统计描述, 找出不表达的基因> summary(data["A9QA",])> names(data3["A9QA",which(data3["A9QA",]==0)]) ; names(向量)> colnames(data3[,which(data3["A9QA",]==0)]) ; colnames(矩阵)#从数据中随机选取 10 个基因得到新的表达矩阵,并绘制箱线图;> samp <- data3[,sample(1:ncol(data3),10)] ; sample取随机数> boxplot(samp)
4.大样本比例检验,故对答案存疑!
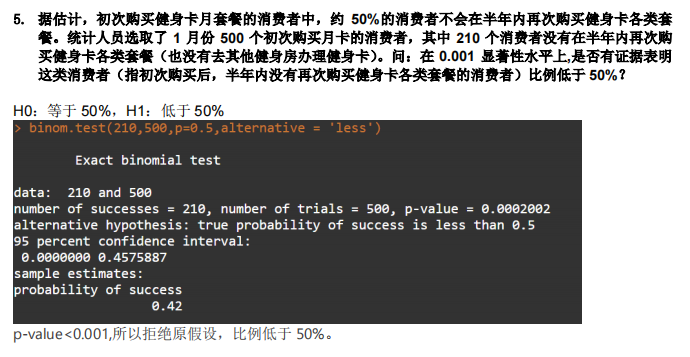
> z <- (210/500 - 0.5) / sqrt(0.25/500) ;z[1] -3.577709> pnorm(z)[1] 0.0001733097因p-value值<0.001,故拒绝原假设,比例低于50%
t检验
> time <- c(4.21, 5.55, 3.02, 5.13, 4.77, 2.34, 3.54, 3.2, 4.5, 6.1, 0.38, 5.12, 6.19, 3.79,1.08)> shapiro.test(time) #小样本要进行正态性检验> t.test(time,alternative = "less",mu = 5)
第四次作业
1.秩检验
> wilcox.test(a,b,paired = FALSE,alternative = "two.sided",exact = FALSE)
2.方差相等的t检验
#检验方差是否同质> var.test(a,b)#t检验> shapiro.test(a)> shapiro.test(b)> t.test(a,b,paired = FALSE,var.equal = TRUE)
3.单样本t-test【step-by-step】
H0:μ=4.15;Ha:μ≠4.15> t <- (4.98-4.15)/(2.78/sqrt(16));t[1] 1.194245> p <- 2*(1-pt(t,15));p[1] 0.2509263因p>0.05,故接收原假设,即认为陈旧性心肌梗死患者的血浆载脂蛋白E平均浓度与正常人无显著差别有统计学意义> left <- 4.98 - qt(0.975,15)*2.78/sqrt(16);left[1] 3.498643> right <- 4.98 + qt(0.975,15)*2.78/sqrt(16);right[1] 6.46135795%CI=[3.498643,6.461357]
第五次作业
1.ANOVA(step-by-step)
#读取数据> group1<-c(243, 251, 275, 291, 347, 354, 380, 392)> group2<-c(206, 210, 226, 249, 255, 273, 285, 295, 309)> group3<-c(241, 258, 270, 293, 328)> folic_acid <- data.frame(value=c(group1,group2,group3),cls=c(rep("G1",length(group1)),rep("G2",length(group2)),rep("G3",length(group3))))> folic_acid <- transform(folic_acid,cls=factor(cls,levels = c("G1","G2","G3")))> attach(folic_acid)#正态性检验> shapiro.test(group1)> shapiro.test(group2)> shapiro.test(group3)#方差齐性检验> bartlett.test(value~cls,folic_acid)#step-by-step#计算平方和> GrandMean <- mean(folic_acid$value);GrandMean> SMeans <- aggregate(folic_acid$value,by=list(folic_acid$cls),FUN=mean);SMeans> SVars <- aggregate(folic_acid$value,by=list(folic_acid$cls),FUN=var);SVars> SLens <- aggregate(folic_acid$value,by=list(folic_acid$cls),FUN=length);SLens> within_SS <- sum((SLens$x-1)*SVars$x)> total_SS <- sum((value-GrandMean)^2)> between_SS <- total_SS-within_SS> df_between <- length(levels(cls))-1> df_within <- length(value) - length(levels(cls))> between_MS <- between_SS/df_between> within_MS <- within_SS/df_within> F_ration <- between_MS/within_MS> P_value <- 1-pf(F_ration,df_between,df_within);P_value[1] 0.04358933因p_value<0.05,故拒绝原假设,即认为三组之间存在显著性差异
2.kruskal.test(R)(序数因变量)
注:数据格式转换 ```r
drug_data <- read.table(“D:/郭东灵/学习/生统/第五次/homework5_data2.csv”,header=TRUE,sep=”,”) View(drug_data) drug_data <-melt(drug_data) attach(drug_data) kruskal.test(value~variable,data=drug_data)
Kruskal-Wallis rank sum test
data: value by variable Kruskal-Wallis chi-squared = 3.0973, df = 2, p-value = 0.2125
<a name="bZrvV"></a>### 3.单因素方差分析(R)```r> production <- read.table("D:/郭东灵/学习/生统/第五次/homework5_data3.txt",header=TRUE,colClass=c("numeric","factor"))#正态性检验> shapiro.test(production$yield[production$seed==1])> shapiro.test(production$yield[production$seed==2])> shapiro.test(production$yield[production$seed==3])#齐性检验> bartlett.test(yield~seed,data=production)#方差分析> summary(aov(yield~seed,data=production))#后续分析> pairwise.t.test(production$yield,production$seed,p.adjust.method = "fdr")> TukeyHSD(aov(yield~seed,data=production))
4.双因素方差分析
> tooth <- read.table("D:/郭东灵/学习/生统/第五次/homework_5_data4.csv",sep=",",header=TRUE,colClass=c("numeric","factor","factor"))> summary(tooth$len)Min. 1st Qu. Median Mean 3rd Qu. Max.4.20 13.07 19.25 18.81 25.27 33.90> summary(aov(len~supp*dose,data = tooth))> TukeyHSD(aov(len~supp*dose,data = tooth))
第六次作业
1.多元线性回归分析
(1)
> Y <- c(162,120,223,131,67,167,81,192,116,55,252,232,144,103,212)> X1 <- c(274,180,375,205,86,265,98,330,195,53,430,372,236,157,370)> X2 <- c(2450,3250,3802,2838,2347,3782,3008,2450,2137,2560,4020,4427,2660,2088,2605)> ff <- lm(Y~X1+X2)> summary(ff)Call:lm(formula = Y ~ X1 + X2)Residuals:Min 1Q Median 3Q Max-3.9014 -1.2923 -0.1171 1.6956 3.7601Coefficients:Estimate Std. Error t value Pr(>|t|)(Intercept) 3.984819 2.553039 1.561 0.145X1 0.496767 0.006360 78.104 < 2e-16 ***X2 0.008913 0.001017 8.762 1.46e-06 ***---Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1Residual standard error: 2.287 on 12 degrees of freedomMultiple R-squared: 0.9988, Adjusted R-squared: 0.9986F-statistic: 5142 on 2 and 12 DF, p-value: < 2.2e-16
由此可得,Y =3.984819 + 0.496767X1 + 0.008913X2
从结果可得,F检验的p-value< 2.2e-16,且对X1和X2的t检验的p-value也都均显著,表明获得的线性回归方程有效
(2)
> newdata <- data.frame(x1=200,x2=3000)> predict(ff,newdata = newdata,interval = "prediction",level = 0.95)fit lwr upr1 130.0772 124.8931 135.2612
当X1=200,X2=3000时,根据预测的回归方程可得Y为130.0772
2.逻辑回归
(1)读数据
> install.packages("ISLR")> library(ISLR)> summary(Default)default student balance incomeNo :9667 No :7056 Min. : 0.0 Min. : 772Yes: 333 Yes:2944 1st Qu.: 481.7 1st Qu.:21340Median : 823.6 Median :34553Mean : 835.4 Mean :335173rd Qu.:1166.3 3rd Qu.:43808Max. :2654.3 Max. :73554> View(Default)
该数据集中共有4个观测值。
(2)逻辑模型
> Default_training <- Default[1:7000,]> fit_tr <- glm(default~student+balance+income,data = Default_training,family = binomial())> summary(fit_tr)Call:glm(formula = default ~ student + balance + income, family = binomial(),data = Default_training)Deviance Residuals:Min 1Q Median 3Q Max-2.1872 -0.1417 -0.0549 -0.0196 3.6863Coefficients:Estimate Std. Error z value Pr(>|z|)(Intercept) -1.101e+01 5.889e-01 -18.704 <2e-16 ***studentYes -6.464e-01 2.846e-01 -2.271 0.0231 *balance 5.829e-03 2.781e-04 20.958 <2e-16 ***income 4.711e-06 9.875e-06 0.477 0.6333---Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1(Dispersion parameter for binomial family taken to be 1)Null deviance: 2090.7 on 6999 degrees of freedomResidual deviance: 1109.4 on 6996 degrees of freedomAIC: 1117.4Number of Fisher Scoring iterations: 8
(3)简化模型
> fit_red <- glm(default~student+balance,data = Default_training,family = binomial())> summary(fit_red)Call:glm(formula = default ~ student + balance, family = binomial(),data = Default_training)Deviance Residuals:Min 1Q Median 3Q Max-2.2079 -0.1420 -0.0550 -0.0196 3.6762Coefficients:Estimate Std. Error z value Pr(>|z|)(Intercept) -1.083e+01 4.405e-01 -24.586 < 2e-16 ***studentYes -7.526e-01 1.763e-01 -4.268 1.97e-05 ***balance 5.832e-03 2.781e-04 20.971 < 2e-16 ***---Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1(Dispersion parameter for binomial family taken to be 1)Null deviance: 2090.7 on 6999 degrees of freedomResidual deviance: 1109.6 on 6997 degrees of freedomAIC: 1115.6Number of Fisher Scoring iterations: 8> anova(fit_red,fit_tr,test = "Chisq")Analysis of Deviance TableModel 1: default ~ student + balanceModel 2: default ~ student + balance + incomeResid. Df Resid. Dev Df Deviance Pr(>Chi)1 6997 1109.62 6996 1109.4 1 0.22749 0.6334
根据检验结果可知,卡方值(p=0.6334)不显著,表明两个模型拟合程度一样好,因此可以使用简化模型来进行解释。
(4)模型有效性
> Default_test <- Default[7001:10000,1:3]> Default_test$prob <- predict(fit_red,newdata = Default_test,type = "response")> summary(Default_test)default student balance probNo :2907 No :2105 Min. : 0.0 Min. :0.0000093Yes: 93 Yes: 895 1st Qu.: 486.1 1st Qu.:0.0002745Median : 826.7 Median :0.0019506Mean : 837.4 Mean :0.03516203rd Qu.:1168.0 3rd Qu.:0.0133454Max. :2654.3 Max. :0.9801269> Default_test <- Default_test[order(Default_test$prob,decreasing = TRUE),]> Default_test[1:93,"prob"] <- "Yes"> Default_test[94:3000,"prob"] <- "No"> Default_test$prob <- factor(Default_test$prob,levels = c("Yes","No"),labels = c("Yes","No"))> Default_test$judg <- ifelse(Default_test$default == Default_test$prob,1,0)> table(Default_test$judg)0 190 2910> accuracy <- 2910/3000*100 ;accuracy[1] 97
故,该训练模型的准确性为97%
3.线性回归
(1)多元线性回归
> crop_yield <- read.table("D:/郭东灵/学习/生统作业/第六次/homework6.3.csv",sep=",",header=TRUE)> fit_crop <- lm(crop_yield~illumination_time+illumination_intensity+chemical_fertilizer,data = crop_yield)> summary(fit_crop)Call:lm(formula = crop_yield ~ illumination_time + illumination_intensity +chemical_fertilizer, data = crop_yield)Residuals:Min 1Q Median 3Q Max-0.41065 -0.11562 -0.00984 0.13466 0.51361Coefficients:Estimate Std. Error t value Pr(>|t|)(Intercept) 7.5891 2.4450 3.104 0.004567 **illumination_time -2.3577 0.6379 -3.696 0.001028 **illumination_intensity 1.6122 0.2954 5.459 1.01e-05 ***chemical_fertilizer 0.5012 0.1259 3.981 0.000491 ***---Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1Residual standard error: 0.2347 on 26 degrees of freedomMultiple R-squared: 0.8936, Adjusted R-squared: 0.8813F-statistic: 72.8 on 3 and 26 DF, p-value: 8.883e-13
(2)模型预测
> testdata$crop_yield <- predict(fit_crop,newdata = testdata)> testdataillumination_time illumination_intensity chemical_fertilizer crop_yield1 0 0 7.77 11.48305
经模型预测,施肥量7.77下黑暗生长的作物产量为11.48305kg
(3)计算R2以及F检验
> crop_yield$pred <- predict(fit_crop,newdata = crop_yield)> Res_SS <- sum((crop_yield$crop_yield - crop_yield$pred)^2) ; Res_SS[1] 1.431813> Total_SS <- sum((crop_yield$crop_yield - mean(crop_yield$crop_yield))^2) ; Total_SS[1] 13.45859> Reg_SS <- Total_SS - Res_SS ; Reg_SS[1] 12.02677> R2 <- Reg_SS/Total_SS ; R2[1] 0.8936135
H0:β1=β2=0 ; Ha:β1、β2中至少有一个不等于0> fit_crop_reduce <- lm(crop_yield~chemical_fertilizer,data = crop_yield)> crop_yield$reduce <- predict(fit_crop_reduce,newdata = crop_yield)> Res_SS_reduce <- sum((crop_yield$crop_yield - crop_yield$reduce)^2) ;Res_SS_reduce[1] 3.131884> Total_SS_reduce <- sum((crop_yield$crop_yield - mean(crop_yield$reduce))^2) ; Total_SS_reduce[1] 13.45859> Reg_SS_reduce <- Total_SS_reduce - Res_SS_reduce ; Reg_SS_reduce[1] 10.3267> Reg_df_reduce <- 2 ;Reg_df_reduce[1] 2> Res_df <- 30-3-1 ; Res_df[1] 26> Reg_MS_reduce <- Reg_SS_reduce / Reg_df_reduce ;Reg_MS_reduce[1] 5.163351> Res_MS <- Res_SS/Res_df ; Res_MS[1] 0.05506972> F <- Reg_MS_reduce / Res_MS ; F[1] 93.76027> 1-pf(F,Reg_df_reduce,Res_df)[1] 1.294076e-12
可知,R2为0.8936135;且F检验的p-value(1.294076e-12)值远<0.01,故拒绝原假设,认为β1、β2中至少有一个不等于0
第七次作业
1.主成分分析
> data(iris)> head(iris, 3)> par(mfrow=c(2,2)) #将画幅进行分割#绘制直方图> hist(iris$Sepal.Length, breaks = 20)> hist(iris$Sepal.Width, breaks = 20)> hist(iris$Petal.Length, breaks = 20)> hist(iris$Petal.Width, breaks = 20)#取对数值> log.iris <- log(iris[,1:4])#主成分分析> iris.pca <- prcomp(log.iris,center = TRUE,scale = TRUE )#center标准分布 #scale量纲标准化> print(iris.pca)> summary(iris.pca)故,需要PC1,PC2
2.聚类分析
> iris.hc <- hclust(dist(iris[,1:4]))> plot(iris.hc,hang = -1)> re <- rect.hclust(iris.hc, k=3)#标注3类> iris.id <- cutree(iris.hc, 3)#分类编号> table(iris.id,iris$Species)iris.id setosa versicolor virginica1 50 0 02 0 23 493 0 27 1
3.RNA-seq
> gene_data <- read.table("D:/郭东灵/学习/生统/第七次/homework7_data2.txt",header=TRUE)#画密度图>library(ggplot2)>library(ggpubr)> pl1 <- plot(density(gene_data$norm1), main="norm1")> plot1 <- ggplot(gene_data,aes(x=norm1)) + geom_density(fill="cornflowerblue",color="white",alpha=0.7) + xlim(-5,50);plot1#画箱线图> pl <- boxplot(gene_data)#求最大最小值> row.names(gene_data)[order(gene_data$sumtumor,decreasing = TRUE)[1]][1] "652991"> row.names(gene_data)[order(gene_data$sumnorm,decreasing=TRUE)[1]][1] "7223"
#limma包得安装> install.packages("BiocManager")> BiocManager::install("limma")> library(limma)#标准化> gene_normlize <- normalizeQuantiles(gene_data[,1:6])> boxplot(gene_normlize)#取对数> gene_log <- log(gene_normlize[,1:6] + 1)> boxplot(gene_log)
#求p值> gene_log$pvalue <- apply(gene_log,1,function(x){+ if(var.test(as.numeric(x[1:3]),as.numeric(x[4:6]))$p.value>0.05)+ t.test(as.numeric(x[1:3]),as.numeric(x[4:6]),alternative="less",var.equal=TRUE)$p.value+ else t.test(as.numeric(x[1:3]),as.numeric(x[4:6]),alternative="less",var.equal=FALSE)$p.value})#求foldchange> gene_log$foldchange <- apply(gene_log[,1:6],1,function(x){(mean(x[4:6])-mean(x[1:3]))/min(mean(x[4:6]),mean(x[1:3]))})#根据条件取值> up_gene <- row.names(gene_log)[which(gene_log$pvalue<0.05 & gene_log$foldchange > 1.5)]> length(up_gene)
#画热图> library(pheatmap)> up_gene_matrix <- gene_log[which(gene_log$pvalue<0.05&gene_log$foldchange>1.5),1:6]> pheatmap(up_gene_matrix)
#fisher算法> kegg_list <- read.table("D:/郭东灵/学习/生统/第七次/homework7_kegg.txt")#计算基因个数> up_in_list <- length(intersect(up_gene,kegg_list))> up_out_list <- length(up_gene) - up_in_list> all_except_up_in_list <- length(intersect(row.names(gene_data),kegg_list))> all_except_up_out_list <- dim(gene_data)[1] - up_in_list - up_out_list - all_except_up_in_list#制作表格> rnames <- c("In anno group","Not in anno group")> cnames <- c("Gene list","Genome")> counts <- matrix(c(up_in_list,up_out_list,all_except_up_in_list,all_except_up_out_list),nrow = 2,dimnames = list(rnames,cnames));countsGene list GenomeIn anno group 2 13Not in anno group 448 5899# 进行Fisher精确检验# H0:列联表中行列变量相互独立# HA:行列变量相互关联> fisher.test(counts,alternative = "greater")Fisher's Exact Test for Count Datadata: countsp-value = 0.2873alternative hypothesis: true odds ratio is greater than 195 percent confidence interval:0.3259235 Infsample estimates:odds ratio2.025459因p-value>0.05,故接受原假设
2020年生统作业
第三次作业
1.R的基本命令
2.分布值计算
#二项分布检验#计算对应的概率密度值>pbinom(q, size, prob, lower.tail = TRUE)#某值的概率dbinom(x, size, prob, log = FALSE)#概率密度pbinom(q, size, prob, lower.tail = TRUE, log.p = FALSE)#计算某概率下的值>qbinom(p, size, prob, lower.tail = TRUE, log.p = FALSE)#二项分布检验>binom.test(x, n, p = 0.5,alternative = c("two.sided", "less", "greater"), conf.level = 0.95)
#正态分布#计算对应z值的概率密度(Pr)>pnorm(q, lower.tail = TRUE)#计算z值>qnorm(p,lower.tail = TRUE)#生成符合某正态分布的随机数>rnorm(n, mean = 0, sd = 1)
3.单个个体的正态检验:手动计算
> mu <- 200> sd <- 16> n <- 50> z <- (203.5-mu)/(sd/sqrt(n));z[1] 1.546796> p <- 2*(1-pnorm(z));p[1] 0.1219124
4.读入文件,绘值箱线图
#读入文件> gene_pre <- read.csv("D:/郭东灵/学习/生统/题目/19级作业答案/biostatistic_homework/第三次作业/homework3-4_data.csv",header=TRUE,row.names=1,sep=",") #设置行名;#标记列名#绘制密度图> plot(density(as.numeric(gene_pre[3,]))) #将列表中的数据作为输入时,需要as.numeric将数据转为数字格式#导出pdf文件> pdf("G3_density_plot.pdf")> plot(density(as.numeric(gene_pre[3,])))> dev.off()RStudioGD2#计算最小值、中位数和方差> median(as.numeric(gene_pre[3,]))[1] 10.08245> var(as.numeric(gene_pre[3,]))[1] 0.3880644> min(as.numeric(gene_pre[3,]))[1] 9.271463#去掉离群值> boxplot(gene_pre)> gene_pre_new <- gene_pre> gene_pre_new[gene_pre_new>100] <- NA> boxplot(gene_pre_new)#只在作图时不看离群值> boxplot(gene_pre,outline = FALSE)
第四次作业
1.手动t检验及置信区间
> t <- (4.98-4.15)/(2.78/sqrt(16));t[1] 1.194245> p <- 2*(1-pt(t,15));p[1] 0.2509263> left <- 4.98 - qt(0.975,15)*2.78/sqrt(16);left[1] 3.498643> right <- 4.98 - qt(0.975,15)*2.78/sqrt(16);right[1] 3.498643> right <- 4.98 + qt(0.975,15)*2.78/sqrt(16);right[1] 6.461357
2.秩和检验
> a <- c(113,120,138,120,100,118,138,123)> b <- c(138,116,125,136,110,132,130,110)> wilcox.test(a,b,paired = FALSE,exact = FALSE)
3.标准、完整的t检验
#先做假设#正态检验> shapiro.test(keshan$patient)Shapiro-Wilk normality testdata: keshan$patientW = 0.97286, p-value = 0.4412> shapiro.test(keshan$healthy)Shapiro-Wilk normality testdata: keshan$healthyW = 0.97239, p-value = 0.427#方差齐性检验> var.test(keshan[,1],keshan[,2])F test to compare two variancesdata: keshan[, 1] and keshan[, 2]F = 1.3064, num df = 39, denom df = 39, p-value = 0.4077alternative hypothesis: true ratio of variances is not equal to 195 percent confidence interval:0.6909357 2.4699694sample estimates:ratio of variances1.306365#t检验> t.test(keshan[,1],keshan[,2],var.equal = TRUE)Two Sample t-testdata: keshan[, 1] and keshan[, 2]t = 17.992, df = 78, p-value < 2.2e-16alternative hypothesis: true difference in means is not equal to 095 percent confidence interval:1.333801 1.665699sample estimates:mean of x mean of y5.00625 3.50650
4.t检验
#配对t检验> t.test(BMI2,BMI1,paired = TRUE,alternative = "less")Paired t-testdata: BMI2 and BMI1t = -17.72, df = 9, p-value = 1.316e-08alternative hypothesis: true difference in means is less than 095 percent confidence interval:-Inf -1.394139sample estimates:mean of the differences-1.555#单样本t检验> t.test(BMI2,mu=23.2,alternative = "greater")One Sample t-testdata: BMI2t = 7.0921, df = 9, p-value = 2.858e-05alternative hypothesis: true mean is greater than 23.295 percent confidence interval:24.61632 Infsample estimates:mean of x25.11#计算样本数量>library(pwr)> pwr.t.test(d = d,sig.level = 0.001,type = "one.sample",alternative = "greater",power = 0.95) #注意power的含义为1-typeⅡ error #d的含义为effect sizeOne-sample t test power calculationn = 9.181621d = 2.242734sig.level = 0.001power = 0.95alternative = greater
5.多假设检验矫正
> snp <- read.csv("D:/郭东灵/学习/生统/题目/19级作业答案/biostatistic_homework/第四次作业/homework4-5_data.csv",header=TRUE)> snp$bonfer <- p.adjust(snp$p.value,method = "bonferroni",length(snp$p.value))> snp$SNP[snp$bonfer<0.05][1] 7> snp$fdr <- p.adjust(snp$p.value,method = "fdr",length(snp$p.value))> snp$SNP[snp$fdr<0.05][1] 7 8 10故可知,fdr的矫正结果比bonferroni矫正更为宽松
6.t检验
> expression_data <- read.table("D:/郭东灵/学习/生统/题目/19级作业答案/biostatistic_homework/第四次作业/homework4-6_data.txt",header=TRUE,row.names=1)> expression_data$pvalue <- apply(expression_data,1,function(x){+ if(var.test(as.numeric(x[1:40]),as.numeric(x[41:80]))$p.value>0.05)+ t.test(as.numeric(x[1:40]),as.numeric(x[41:80]),var.equal = TRUE)$p.value+ else t.test(as.numeric(x[1:40]),as.numeric(x[41:80]),var.equal = FALSE)$p.value})> rownames(expression_data[which(expression_data$pvalue>0.05),])[1] "gene_7"故可知,gene7在两组间不存在显著性差异1> expression_data$bonferr <- p.adjust(expression_data$pvalue,method = "bonferroni")> rownames(expression_data[which(expression_data$bonferr>0.05),])[1] "gene_1" "gene_2" "gene_3" "gene_7" "gene_8" "gene_10" "gene_15" "gene_17" "gene_19"> expression_data$fdr <- p.adjust(expression_data$pvalue,method = "fdr")> rownames(expression_data[which(expression_data$fdr>0.05),])[1] "gene_7"
第五次作业
1.ANOVA单因素step-by-step+方差不等的韦尔奇检验
> alt <- read.csv("D:/郭东灵/学习/生统/题目/19级作业答案/biostatistic_homework/第五次作业/data5_3.csv",header=TRUE)> boxplot(alt)H0:ABC之间不存在显著性差异;Ha:至少有一组存在显著性差异#格式转变> alt_trans <- data.frame(alt=c(alt$A,alt$B,alt$C),effect=c(rep("A",15),rep("B",15),rep("C",15)))> alt_trans <- transform(alt_trans,effect=factor(effect,levels = c("A","B","C")))#正态性检验> shapiro.test(alt$A)> shapiro.test(alt$B)> shapiro.test(alt$C)#方差齐性检验> bartlett.test(alt~effect,alt_trans)#韦尔奇step-by-step> GrandMean <- mean(alt_trans$alt);GrandMean[1] 16.80667> SMeans <- aggregate(alt_trans$alt,by=list(alt_trans$effect),FUN=mean);SMeans> SVars <- aggregate(alt_trans$alt,by=list(alt_trans$effect),FUN=var);SVars> SLens <- aggregate(alt_trans$alt,by=list(alt_trans$effect),FUN=length);SLens> within_SS <- sum((SLens$x-1)*SVars$x)> total_SS <- sum((alt_trans$alt-GrandMean)^2)> between_SS <- total_SS-within_SS> df_between <- length(levels(alt_trans$effect))-1> df_within <- length(alt_trans$alt) - length(levels(alt_trans$effect))> between_MS <- between_SS/df_between;between_MS[1] 1197.954> within_MS <- within_SS/df_within;within_MS[1] 21.06286> k <- length(levels(alt_trans$effect));k[1] 3> n.i <- SLens$x;n.i[1] 15 15 15> m.i <- SMeans$x;m.i[1] 7.346667 25.106667 17.966667> v.i <- SVars$x;v.i[1] 5.406952 46.850667 10.930952> w.i <- n.i/v.i;w.i[1] 2.7742060 0.3201662 1.3722501> sum.w.i <- sum(w.i);sum.w.i[1] 4.466622> tmp <- sum((1-w.i/sum.w.i)^2/(n.i-1))/(k^2-1);tmp[1] 0.01326148> m <- sum(w.i * m.i)/sum.w.i;m[1] 11.88241> F_welch <- sum(w.i * (m.i-m)^2)/((k-1)*(1+2*(k-2)*tmp));F_welch[1] 79.8145> pvalue_welch <- pf(F_welch,k-1,1/(3*tmp),lower.tail = FALSE);pvalue_welch[1] 1.294731e-11#R代码计算> oneway.test(alt~effect,data=alt_trans,var.equal = FALSE)One-way analysis of means (not assuming equal variances)data: alt and effectF = 79.814, num df = 2.000, denom df = 25.135, p-value = 1.295e-11#后续多重比较> pairwise.t.test(alt_trans$alt,alt_trans$effect,p.adjust.method = "bonferroni")Pairwise comparisons using t tests with pooled SDdata: alt_trans$alt and alt_trans$effectA BB 5.8e-13 -C 3.9e-07 0.00034P value adjustment method: bonferroni故可知,三个样本之间均存在显著性差异
2.双因素方差分析
> data2 <- read.csv("D:/郭东灵/学习/生统/题目/19级作业答案/biostatistic_homework/第五次作业/data5_4.csv",header=TRUE)> data2 <- transform(data2,A=factor(A,levels = c("A1","A2","A3")),B=factor(B,levels = c("B1","B2")))> shapiro.test(data2$Weight[data2$A=="A1"])> shapiro.test(data2$Weight[data2$A=="A2"])> shapiro.test(data2$Weight[data2$A=="A3"])> shapiro.test(data2$Weight[data2$B=="B1"])> shapiro.test(data2$Weight[data2$B=="B2"])> bartlett.test(Weight~A,data = data2)> bartlett.test(Weight~B,data = data2)#R包直接分析> summary(aov(Weight~A*B,data = data2))> interaction.plot(data2$A,data2$B,data2$Weight,type = "b",col = c("red","blue"),pch = c(16,18),main = "Interaction between A and B")> library(gplots)> plotmeans(data2$Weight~interaction(data2$A,data2$B,sep=" "),connect = list(c(1,2),c(3,4)),col = c("red","blue"),main="plot with 95% CIs",xlab = "age and sex combination")#双因素方差分析step-by-step> GrandMean <- mean(data2$Weight);GrandMean> SMeansA <- aggregate(data2$Weight,by=list(data2$A),FUN=mean);SMeansA> SMeansB <- aggregate(data2$Weight,by=list(data2$B),FUN=mean);SMeansB> SMeansAB <- aggregate(data2$Weight,by=list(data2$A,data2$B),FUN=mean);SMeansAB> SVarsA <- aggregate(data2$Weight,by=list(data2$A),FUN=var);SVarsA> SVarsB <- aggregate(data2$Weight,by=list(data2$B),FUN=var);SVarsB> SVarsAB <- aggregate(data2$Weight,by=list(data2$A,data2$B),FUN=var);SVarsAB> SLenA <- aggregate(data2$Weight,by=list(data2$A),FUN=length);SLenA> SLenB <- aggregate(data2$Weight,by=list(data2$B),FUN=length);SLenB> SLenAB <- aggregate(data2$Weight,by=list(data2$A,data2$B),FUN=length);SLenAB> withinSSA <- sum((SLenA$x-1)*SVarsA$x);withinSSA> betweenSSA <- sum(SLenA$x*SMeansA$x^2)-length(data2$A)*(GrandMean)^2;betweenSSA> withinSSB <- sum((SLenB$x-1)*SVarsB$x);withinSSB> betweenSSB <- sum(SLenB$x*SMeansB$x^2)-length(data2$B)*(GrandMean)^2;betweenSSB> SSE <- sum(data2$Weight^2)-sum(SMeansAB$x^2*SLenAB$x);SSE> SSTotal <- sum((data2$Weight-GrandMean)^2);SSTotal> betweenSSAB <- SSTotal - betweenSSA - betweenSSB - SSE;betweenSSAB> n <- 10> df_betweenA <- length(levels(data2$A))-1;df_betweenA[1] 2> df_betweenB <- length(levels(data2$B))-1;df_betweenB[1] 1> df_betweenAB <- df_betweenA*df_betweenB;df_betweenAB[1] 2> df_E <- length(levels(data2$A)) * length(levels(data2$B)) * (n-1);df_E[1] 54> df_total <- length(levels(data2$A)) * length(levels(data2$B)) * n-1;df_total[1] 59> MSA <- betweenSSA/df_betweenA;MSA[1] 4540.214> MSB <- betweenSSB/df_betweenB;MSB[1] 127.0215> MSAB <- betweenSSAB/df_betweenAB;MSAB[1] 8.664> MSE <- SSE/df_E;MSE[1] 79.92094> FA <- MSA/MSE;FA[1] 56.80881> FB <- MSB/MSE;FB[1] 1.589339> FAB <- MSAB/MSE;FAB[1] 0.1084071> p_value_A <- pf(FA,df_betweenA,df_E,lower.tail = FALSE);p_value_A[1] 5.223962e-14> p_value_B <- pf(FB,df_betweenB,df_E,lower.tail = FALSE);p_value_B[1] 0.2128406> p_value_AB <- pf(FAB,df_betweenAB,df_E,lower.tail = FALSE);p_value_AB[1] 0.897457#后续多重分析> TukeyHSD(aov(Weight~A*B,data = data2))> p_adj <- TukeyHSD(aov(Weight~A*B,data = data2))$`A:B`[TukeyHSD(aov(Weight~A*B,data = data2))$`A:B`[,"p adj"]<0.05,]> rownames(p_adj)[grep(rownames(p_adj),pattern = "A2:B1")][1] "A2:B1-A1:B1" "A1:B2-A2:B1"
3.单因素方差分析
> data3 <- read.csv("D:/郭东灵/学习/生统/题目/19级作业答案/biostatistic_homework/第五次作业/protein.csv",header=TRUE,row.names=1)> data3_standardlized <- apply(data3,2,function(x){x/mean(x)})> boxplot(log10(data3_standardlized))#按行进行多因素方差分析> groups <- rep(c("A","B","C"),each=5)aov_per_gen <- apply(data3_standardlized,1,function(x){unlist(summary(aov(x~factor(groups))))["Pr(>F)1"]})> length(aov_per_gen[aov_per_gen<0.05])[1] 1048> anova_per_gene <- apply(data3_standardlized,1,function(x){anova(lm(x~factor(groups)))$'Pr(>F)'[1]})> length(anova_per_gene[anova_per_gene<0.05])[1] 1048#多假设检验矫正> anova_per_gene_padjust <- p.adjust(anova_per_gene,method = "bonferroni")> length(anova_per_gene_padjust[anova_per_gene_padjust <0.05])[1] 203
第六次作业
1.回归分析
> data(cars)> View(cars)> head(cars,6)speed dist1 4 22 4 103 7 44 7 225 8 166 9 10#绘制散点图并拟合平滑的曲线> scatter.smooth(x=cars$speed,y=cars$dist,main="Dist~Speed")#绘制密度图以及检验正态分布> plot(density(cars$speed))> plot(density(cars$dist))> shapiro.test(cars$speed)Shapiro-Wilk normality testdata: cars$speedW = 0.97765, p-value = 0.4576> shapiro.test(cars$dist)Shapiro-Wilk normality testdata: cars$distW = 0.95144, p-value = 0.0391#计算回归系数,构建线性模型> cor(cars$speed,cars$dist)[1] 0.8068949设回归模型为 y=α+βx> linearMode <- lm(dist~speed,data=cars)> summary(linearMode)Call:lm(formula = dist ~ speed, data = cars)Residuals:Min 1Q Median 3Q Max-29.069 -9.525 -2.272 9.215 43.201Coefficients:Estimate Std. Error t value Pr(>|t|)(Intercept) -17.5791 6.7584 -2.601 0.0123 *speed 3.9324 0.4155 9.464 1.49e-12 ***---Signif. codes:0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1Residual standard error: 15.38 on 48 degrees of freedomMultiple R-squared: 0.6511, Adjusted R-squared: 0.6438F-statistic: 89.57 on 1 and 48 DF, p-value: 1.49e-12故回归方程为 y=-17.5791+3.9324x有 Pr(>|t|) 值可知,统计模型显著
2.多元+二项回归分析
> install.packages("mlbench")> library(mlbench)> data(BreastCancer)> bc <- BreastCancer[complete.cases(BreastCancer), ] #create copy,避免更改原始数据集#数据结构特征> str(bc)#将因子装换为数字> bc <- BreastCancer[complete.cases(BreastCancer), ]> for (i in 2:10) {+ bc[,i] <- as.numeric(as.character(bc[,i]))+ }#编码二元响应变量,并设为因子> bc$Class <- ifelse(bc$Class=="malignant",1,0)> bc$Class <- factor(bc$Class,levels = c(0,1))#二元变量回归分析 #数据必须设置为数字> f2full <- glm(Class~Cl.thickness+Cell.size+Cell.shape+Marg.adhesion+Epith.c.size+Bare.nuclei+Bl.cromatin+Normal.nucleoli+Mitoses,data = bc,family = binomial())> summary(f2full)#简化模型#答题时要注意写设拟合回归方程为logit(P)=ln(odds)=ln(P/(1-P))=α+∑5(i=1) βixi> f2reduce <- glm(Class~Cl.thickness+Marg.adhesion+Bare.nuclei+Bl.cromatin+Normal.nucleoli,data = bc,family = binomial())> summary(f2reduce)#两个模型假设检验H0: 两模型没有显著差异 H1: 两模型有显著差异> anova(f2reduce,f2full,test = "Chisq")因为p_value>0.05,故没有显著性差异#模型训练,计算准确性> training <- bc[1:400,]> testing <- bc[401:683,]> f2training <- glm(Class~Cl.thickness+Marg.adhesion+Bare.nuclei+Bl.cromatin+Normal.nucleoli,data = training,family = binomial())> summary(f2training)#根据模型计算> p1 <- predict(f2training,newdata = testing,type = "response")#计算准确率> n1 <- length(intersect(which(p1<0.5),which(testing$Class=="0")))> n2 <- length(intersect(which(p1>=0.5),which(testing$Class=="1")))> prob <- (n1+n2)/dim(testing)[1];prob[1] 0.9858657
4.step-by-step计算
#回归分析> fev <- read.table("D:/BaiduNetdiskWorkspace/生统复习/生统/题目/19级作业答案/biostatistic_homework/第六次作业/FEV.DAT.txt",header=TRUE)> f3 <- lm(FEV~Age+Hgt+Sex+Smoke,data = fev)> summary(f3)#计算预测值#先创建数据表格> nd <- data.frame(Age=15,Hgt=65,Sex=1,Smoke=0)#进行预测(与二元的预测不同)> predict(f3,newdata = nd,interval = "prediction",level = 0.95)fit lwr upr1 3.455732 2.641431 4.270032#计算R2(R2是总体模型的预测)#预测值> fev$fev_pre <- predict(f3,newdata = fev,interval = "prediction",level = 0.95)[,1]#计算> Res_SS <- sum((fev$FEV-fev$fev_pre)^2);Res_SS[1] 110.2796> Total_SS <- sum((fev$FEV-mean(fev$FEV) )^2);Total_SS[1] 490.9198> Reg_SS <- Total_SS - Res_SS ; Reg_SS[1] 380.6403> R2 <- Reg_SS/Total_SS ; R2[1] 0.7753614#F检验(不区分参数的个数,总体检验)> Reg_df <- 4> Res_df <- length(fev$FEV)-Reg_df-1> F <- (Reg_SS/Reg_df)/(Res_SS/Res_df);F[1] 560.0212> pf(F,Reg_df,Res_df,lower.tail = FALSE)[1] 9.101982e-209
第七次作业
1.经典聚类分析
> cll <- read.table("D:/BaiduNetdiskWorkspace/生统复习/生统/题目/19级作业答案/biostatistic_homework/第七次作业/CLL_expr.txt",header=TRUE)#标准化> library(limma)> cll_normlize <- normalizeQuantiles(cll)#箱线图> boxplot(cll)> boxplot(cll_normlize)#求p——value> DEG_fdr <- apply(cll_normlize , 1, function(x)+ p.adjust(wilcox.test(x[seq(1,11,2)],x[seq(2,12,2)],paired=TRUE,exact=FALSE)$p.value,method = "fdr"))#求foldchange> DEG_fc <- apply(cll_normlize , 1, function(x){abs(mean(x[seq(1,11,2)])-mean(x[seq(2,12,2)]))/min(mean(x[seq(1,11,2)]),mean(x[seq(2,12,2)]))})#寻找交集> DEG <- intersect(names(DEG_fdr[DEG_fdr<0.05]),names(DEG_fc[DEG_fc>2]))
> library(pheatmap)> cll_DEG <- cll_normlize[rownames(cll_normlize) %in% DEG,]#聚类+绘制热图> pheatmap(cll_DEG,annotation = data.frame(cll_group$Subclone,row.names = colnames(cll_DEG)))#GO估计分析——fisher> go <- read.csv("D:/BaiduNetdiskWorkspace/生统复习/生统/题目/19级作业答案/biostatistic_homework/第七次作业/GO0008528.csv",header=TRUE)> go_in_genome <- intersect(go$gene_symbol,rownames(cll_normlize))> in_list <- length(intersect(DEG,go_in_genome));in_list[1] 3> out_list <- length(DEG)-in_list;out_list[1] 31> all_except_in_list <- length(intersect(go$gene_symbol,rownames(cll_normlize)));all_except_in_list[1] 124> all_except_in_list <- length(intersect(go$gene_symbol,rownames(cll_normlize)))-in_list;all_except_in_list[1] 121> all_except_out_list <- length(rownames(cll_normlize))-in_list - out_list - all_except_in_list;all_except_out_list[1] 13506> rnames <- c("In anno group","Not in anno group")> cnames <- c("Gene list","Genome")> counts <- matrix(c(in_list,out_list,all_except_in_list,all_except_out_list),nrow=2,dimnames=list(rnames,cnames));counts#fisher检验> fisher.test(counts,alternative = "greater")
2.PCA主成分分析
> stock <- read.table("D:/BaiduNetdiskWorkspace/生统复习/生统/题目/19级作业答案/biostatistic_homework/第七次作业/stocks.txt",header=TRUE,row.names=1)> stock_pca <- prcomp(stock,center = TRUE,scale. = TRUE)> summary(stock_pca)由此可知,PC1解释了53.6%的variability , PC2解释了21%的variability。根据Cumulative Proportion,可以知道,需要保留PC1-PC4。#biplot图> biplot(stock_pca)> print(stock_pca)由此可以看出,PC1主要由entry_price, market_cap, cash_and_marketable, tev, revenues, ebitda组成。
2019年生统作业
第三次作业
1.t检验
#正态性检验> sup_1<- c(6802,5730,5823,5915,5774,5880,5870,5773,5830,5841,5763,5851,5789,5796,5818,5685,5602, 5841,5723,5757)> sup_2<- c(5884,5871,5797,5957,5803,5862,5814,5885,5856,5940,5945,5803,5864,5851,5714,5943,5830, 5858,5922,5866)> shapiro.test(sup_1)> shapiro.test(sup_2)sup_1不符合正态分布,但是题目规定进行t检验#方差齐性检验> var.test(sup_1,sup_2,alternative = "two.sided")#t检验> t.test(sup_1,sup_2,alternative = "two.sided",var.equal = FALSE)可得p_value>0.05,不能拒绝原假设,即认为两种灯泡的使用寿命时间无显著性差别
2.分布值计算
#正态分布值计算> pnorm(2,mean = 4,sd=1)[1] 0.02275013> qnorm(0.05,mean = 4,sd=1,lower.tail = FALSE)[1] 5.644854
#二项分布值计算> dbinom(0,8,0.5)[1] 0.00390625> dbinom(4,8,0.5)[1] 0.2734375> 1-pbinom(5,8,0.5)[1] 0.1445312> pbinom(5,8,0.5,lower.tail = FALSE)[1] 0.1445313
3.绘制箱线图和柱形图
#产生符合正态分布的数据> n1000 <- rnorm(1000,mean = 100,sd=8)> hist(n1000)> boxplot(n1000)
4.置信区间、检验效力等的手动计算
故需要对其计算原理和步骤熟悉,并做好笔记
第四次作业
1.标准t检验流程
2.配对双样本秩和检验
3.单样本的秩和检验
> insurance <- c(4152,4579,5053,5112,5745,6250,7081,9048,12095,14430,17220,20610,22836,48950,67200)> wilcox.test(insurance,mu=7520)
4.配对t检验
# 导入数据> expression_data <- read.table('./data/Data.txt',header = T,stringsAsFactors =F )> View(expression_data) ##观察数据可发现前十列为control组,后十列为case组#计算p值> t.test.p.value <- function(x){ p_value <- t.test(x[1:10],x[11:20],paired = TRUE)$p.value p_value }> p.t.test <- apply(expression_data,1,t.test.p.value)#显著差异表达的基因数> length(p.t.test[p.t.test<0.05])#前10个差异表达的基因> names(p.t.test[order(p.t.test,decreasing=F)[1:10]])#多假设检验校正
第五次作业
1.标准的anova分析流程
#导入数据> yield <- read.delim("./data/yield.txt")#正态检验# H0:数据符合正态分布 H1:数据不符合正态分布。> shapiro.test(yield$yield[yield$seed==1])> shapiro.test(yield$yield[yield$seed==2])> shapiro.test(yield$yield[yield$seed==3])#方差齐性检验# H0:数据方差齐性 H1:数据方差非齐性> bartlett.test(yield~seed,data = yield)#单样本多因素方差分析> seed_aov <- aov(yield~factor(seed),data = yield)> summary(seed_aov)#后续差异分析_方法1> tukey_results <- TukeyHSD(seed_aov);tukey_results> plot(tukey_results)#后续差异分析_方法2> pairwise.t.test(yield$yield,yield$seed,p.adjust.method = "none")
2019年考试题目
1.
2.
3.单因素方差分析(step-by-step)
#读取数据> drug <- read.csv("D:/BaiduNetdiskWorkspace/生统复习/生统/题目/19级作业答案/2019年考试题目/考试时用到的数据/q2_data.csv",header=TRUE,colClass =c("numeric","factor"))#方差齐性检验> bartlett.test(Performance_Scores~Treatment,data = drug)#单因素方差分析(step-by-step)H0:不存在显著性差异;Ha:至少有一组存在显著性差异> GrandMean <- mean(drug$Performance_Scores);GrandMean[1] 57.3> SMeans <- aggregate(drug$Performance_Scores,by=list(drug$Treatment),FUN=mean);SMeansGroup.1 x1 anxietyDrug 50.02 excitingDrug 83.43 hypertensionDrug 79.24 Placebos 16.6> SVars <- aggregate(drug$Performance_Scores,by=list(drug$Treatment),FUN=var);SVarsGroup.1 x1 anxietyDrug 109.02 excitingDrug 112.33 hypertensionDrug 150.74 Placebos 112.3> SLens <- aggregate(drug$Performance_Scores,by=list(drug$Treatment),FUN=length);SLensGroup.1 x1 anxietyDrug 52 excitingDrug 53 hypertensionDrug 54 Placebos 5> within_SS <- sum((SLens$x-1)*SVars$x);within_SS[1] 1937.2> total_SS <- sum((drug$Performance_Scores-GrandMean)^2) ;total_SS[1] 16290.2> between_SS <- total_SS-within_SS;between_SS[1] 14353> df_between <- length(levels(drug$Treatment))-1;df_between[1] 3> df_within <- length(drug$Performance_Scores) - length(levels(drug$Treatment));df_within[1] 16> between_MS <- between_SS/df_between;between_MS[1] 4784.333> within_MS <- within_SS/df_within;within_MS[1] 121.075> F_ration <- between_MS/within_MS;F_ration[1] 39.51545> P_value <- 1-pf(F_ration,df_between,df_within);P_value[1] 1.262592e-07因此可知p_value远小于0.05,故拒绝原假设,即认为至少有一组存在差异。#aov包> summary(aov(Performance_Scores~Treatment,data = drug))Df Sum Sq Mean Sq F value Pr(>F)Treatment 3 14353 4784 39.52 1.26e-07 ***Residuals 16 1937 121---Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1> TukeyHSD(aov(Performance_Scores~Treatment,data = drug))
#计算cor,计算相关性> BC$diagnosis <- as.numeric(as.character(BC$diagnosis))> cor(BC$diagnosis,BC[,2:11])radius texture perimeter area[1,] 0.7300285 0.4151853 0.7426355 0.7089838smoothness compactness concavity concave.points[1,] 0.35856 0.5965337 0.6963597 0.7766138symmetry fractal_dimension[1,] 0.3304986 -0.0128376> pairs(diagnosis~radius+texture+perimeter+area+smoothness+compactness+concavity+concave.points+symmetry+fractal_dimension,data = BC)
#训练> training <- BC[1:400,]> testing <- BC[401:,]错误: 意外的',' in "testing <- BC[401:,"> testing <- BC[401:569,]> training$diagnosis <- factor(training$diagnosis,levels = c(0,1))> f3full <- glm(diagnosis~radius+texture+perimeter+area+smoothness+compactness+concavity+concave.points+symmetry+fractal_dimension,data = training,family = binomial())> summary(f3full)#计算准确率> testing$predict <- predict(f3full,newdata = testing,type = "response")> testing$predict <- ifelse(testing$predict >0.5,1,0)> a <- length(rownames(testing[which(testing$diagnosis == testing$predict),]));a[1] 153> accuracy <- a / length(rownames(testing));accuracy[1] 0.9053254
4.基因差异分析
> P_value <- apply(AD[,2:11],2,function(x){if(var.test(x[1:161],x[162:346])$p.value>0.05)+ t.test(x[1:161],x[162:346],var.equal=TRUE)$p.value+ else+ t.test(x[1:161],x[162:346],var.equal=FALSE)$p.value})> p_value_adjest <- p.adjust(P_value,method = "bonferroni");p_value_adjest> names(p_value_adjest[p_value_adjest<0.05])[1] "ABR" "ACTN4" "ADAM8" "ADAMTSL4" "AFF3"[6] "AKAP5" "AP5B1" "APIP" "ARF4" "ARHGAP4"> AD_fcABR ACTN4 ADAM8 ADAMTSL4 AFF30.009096499 0.012365542 0.013486465 0.024721369 0.032628461AKAP5 AP5B1 APIP ARF4 ARHGAP40.030557573 0.015770836 0.037186678 0.008875474 0.015433558虽然存在显著性差异,但基因表达倍数改变不大,故不能跳出其中表达存在显著性差异表达的基因
2015年考试题目
1.t检验
1.均值、方差、箱线图
#分别用小函数去求#学到一个新的函数> library(psych)> describe(q1_15)vars n mean sd median trimmed madreticulyte 1 30 6.86 2.29 7.19 6.91 2.83lymphocyte 2 30 3326.49 1187.68 3448.31 3351.60 1671.88min max range skew kurtosis sereticulyte 3.14 9.99 6.85 -0.19 -1.52 0.42lymphocyte 1396.15 4984.70 3588.55 -0.20 -1.47 216.84
2.方差齐性检验、双样本t检验
3.非参数检验(秩检验)
4.比较参数检验和非参数检验
- 差异:
1)参数检验:以已知分布(如正态分布)为假定条件,对总体参数进行估计或检验。
2)非参数检验:不依赖总体分布的具体形式和检验分布(如位置)是否相同。
- 优缺点:
1)参数检验:优点是符合条件时,检验效率高;其缺点是对资料要求严格,如等级数据、非确定数据(>50mg)不能使用参数检验,而且要求资料的分布型已知和总体方差相等。
2)非参数检验:优点是应用范围广、简便、易掌握;缺点是若对符合参数检验条件的资料用非参数检验,则检验效率低于参数检验。如无效假设是正确的,非参数法与参数法一样好,但如果无效假设是错误的,则非参数检验效果较差,如需检验出同样大小的差异的差异往往需要较多的资料。另一点是非参数检验统计量是近似服从某一部分,检验的界值表也是有近似的(如配对秩和检验)因此其结果有一定近似性。
- 非参数检验适用的情况:
(1)等级顺序资料。
(2)偏态资料。当观察资料呈偏态或极度偏态分布而有未经变量变换,或虽经变量变换但仍未达到正态或近似正态分布时,宜用非参数检验。
(3)未知分布型资料
(4)要比较的各组资料变异度相差较大,方差不齐,且不能变换达到齐性。
(5)初步分析。有些医学资料由于统计工作量过大,可采用非参数统计方法进行初步分析,挑选其中有意义者再进一步分析(包括参数统计内容)
(6)对于一些特殊情况,如从几个总体所获得的数据,往往难以对其原有总体分布作出估计,在这种情况下可用非参数统计方法。
2.线性回归
3.作图
1.密度图.pdf
> pdf("the distributation G3 gene expression among 20 samples.pdf")> plot(density(as.numeric(q3_15[3,])),main="the distributation G3 gene expression among 20 samples")> dev.off()
2.箱线图+离群值
> boxplot(q3_15)> boxplot(q3_15,outline = FALSE)#删除离群值【造表格删除值不同于列表】> for(i in 1:20){ #造表格需要按列操作+ OutVals <- boxplot(q3_15[i])$out #获取离群值+ which(q3_15[[i]] %in% OutVals)+ delete <- q3_15[[i]][! q3_15[[i]] %in% OutVals] #删除离群值+write.table(t(delete),file = "q3_15_new.csv",sep = ",",append = TRUE,col.names = FALSE,row.names = FALSE)} #写入> q3_15_new <- read.csv("q3_15_new.csv") #读取新文件> boxplot(t(q3_15_new))
4.ANOVA分析
1.单因素方差分析
#方差其性检验#计算因变量(要有统一标定)> q4_15$A <- q4_15$exp_A/q4_15$exp_actin#自变量要注意设置为因子> summary(aov(A~as.factor(hospital) ,data = q4_15))
注:进行方差分析是,务必检验自由度是否正确【设置因子】,不要只顾着敲命令,一定要检查输出。
2.逻辑回归
> q4_glm <- glm(as.factor(status)~as.factor(abnormal),data = q4_15[q4_15$hospital == 1,],family = binomial())> summary(q4_glm)
5.假设检验
1.标准化+识别缺失值
> sum(is.na(q5_15_data))[1] 0> library(limma)> q5_15_data_nor <- normalizeQuantiles(q5_15_data)
2.apply做t检验
#t检验> q5_15_data_nor$pvalue <- apply(q5_15_data_nor,1,function(x){+ if(var.test(x[11:18],x[1:10])$p.value >0.05)+ t.test(x[11:18],x[1:10],var.equal=TRUE,alternativate="greater")$p.value+ else+ t.test(x[11:18],x[1:10],var.equal=FALSE,alternativate="greater")$p.value})#列出前20的差异基因> row.names(q5_15_data_nor[order(q5_15_data_nor$pvalue,decreasing = FALSE),]) [1:20]
3.在另一个表中找基因
> q5_15 <- data.frame(q5_15) #注意数据形式,list or data.frame> DEGnames <- rownames(q5_15[q5_15$p.value<0.05])> UNDEGnames <- rownames(UNDEG)[1:length(DEGnames )]> DEG_met <- q5_15[q5_15$V1 %in% DEGnames,]> UNDEG_met <- q5_15[q5_15$V1 %in% UNDEGnames,]> var.test(DEG_met$V2,UNDEG_met$V2)> t.test(DEG_met$V2,UNDEG_met$V2,var.equal = FALSE)
4.检验效力
注:千万注意不同包中d的含义
> pwr.t.test(d=d,n=length(DEGnames),type = "two.sample",alternative = "two.sided")Two-sample t test power calculationn = 2093d = 97158.13sig.level = 0.05power = 1alternative = two.sidedNOTE: n is number in *each* group> ?power.t.test> power.t.test(n=length(DEGnames),delta = abs(mu_DEG - mu_UNDEG ),sd=s,sig.level = 0.95,type = "two.sample",alternative = "two.sided")Two-sample t test power calculationn = 2093delta = 0.08765963sd = 4.127671e-05sig.level = 0.95power = 1alternative = two.sidedNOTE: n is number in *each* group

若有收获,就点个赞吧
0 人点赞
