差异分析后,上下调的基因做富集
#单细胞差异分析后的普通的GO和KEGG注释rm(list = ls())library(Seurat)library(SeuratData)library(ggplot2)library(patchwork)library(dplyr)load(file = 'basic.sce.pbmc.Rdata')DimPlot(pbmc,reduction = 'umap',label = TRUE, pt.size = 0.5) + NoLegend()sce=pbmclevels(Idents(sce))sce = sce[, Idents(sce) %in%c( "FCGR3A+ Mono", "CD14+ Mono" )] # FCGR3A就是CD16levels(Idents(sce))markers_df <- FindMarkers(object = sce,ident.1 = 'FCGR3A+ Mono',ident.2 = 'CD14+ Mono',#logfc.threshold = 0,min.pct = 0.25)head(markers_df)#筛选avg_log2FC > 1cg_markers_df=markers_df[abs(markers_df$avg_log2FC) >1,]dim(cg_markers_df)DoHeatmap(subset(sce, downsample = 150),slot = 'counts',unique(rownames(cg_markers_df)),size=3)#做FCGR3A+ Mono 和CD14+ Mono 之间的差异分析deg=FindMarkers(object = sce,ident.1 = 'FCGR3A+ Mono',ident.2 = 'CD14+ Mono',test.use='MAST') #通常用MAST算法算deghead(deg)save(deg,file = 'deg-by-MAST-for-mono-2-cluster.Rdata')

图 deg
gene_up=rownames(deg[deg$avg_log2FC>0,])
gene_down=rownames(deg[deg$avg_log2FC < 0,])
library(org.Hs.eg.db)
#把SYMBOL转为ENTREZID,这里会损失一部分无法匹配到的
gene_up=as.character(na.omit(AnnotationDbi::select(org.Hs.eg.db,
keys = gene_up,
columns = 'ENTREZID',
keytype = 'SYMBOL')[,2]))
gene_down=as.character(na.omit(AnnotationDbi::select(org.Hs.eg.db,
keys = gene_down,
columns = 'ENTREZID',
keytype = 'SYMBOL')[,2]))
#人家的包就是 entrez ID 设计
library(clusterProfiler)
#function里是kegg和GO以及GSEA分析
source('functions.R')
Function:
## KEGG pathway analysis
### 做KEGG数据集超几何分布检验分析,重点在结果的可视化及生物学意义的理解。
run_kegg <- function(gene_up,gene_down,pro='test'){
library(ggplot2)
gene_up=unique(gene_up)
gene_down=unique(gene_down)
gene_diff=unique(c(gene_up,gene_down))
##over-representation test
#下面把3个基因集分开做超几何分布检验
#首先是上调基因集
kk.up <- enrichKEGG(gene = gene_up,
organism = 'hsa',
#universe = gene_all,
pvalueCutoff = 0.9,
qvalueCutoff =0.9)
head(kk.up)[,1:6]
kk=kk.up
dotplot(kk)
kk=DOSE::setReadable(kk, OrgDb='org.Hs.eg.db',keyType='ENTREZID')
write.csv(kk@result,paste0(pro,'_kk.up.csv'))
#下调基因集
kk.down <- enrichKEGG(gene = gene_down,
organism = 'hsa',
#universe = gene_all,
pvalueCutoff = 0.9,
qvalueCutoff =0.9)
head(kk.down)[,1:6]
kk=kk.down
dotplot(kk)
kk=DOSE::setReadable(kk, OrgDb='org.Hs.eg.db',keyType='ENTREZID')
write.csv(kk@result,paste0(pro,'_kk.down.csv'))
#上下调合并后的基因集
kk.diff <- enrichKEGG(gene = gene_diff,
organism = 'hsa',
pvalueCutoff = 0.05)
head(kk.diff)[,1:6]
kk=kk.diff
dotplot(kk)
kk=DOSE::setReadable(kk, OrgDb='org.Hs.eg.db',keyType='ENTREZID')
write.csv(kk@result,paste0(pro,'_kk.diff.csv'))
kegg_up_dt <- as.data.frame(kk.up)
kegg_down_dt <- as.data.frame(kk.down)
kegg_diff_dt <- as.data.frame(kk.diff)
up_kegg<-kegg_up_dt[kegg_up_dt$pvalue<0.01,];up_kegg$group=1
down_kegg<-kegg_down_dt[kegg_down_dt$pvalue<0.01,];down_kegg$group=-1
#画图设置,这个图很丑,大家可以自行修改
g_kegg=kegg_plot(up_kegg,down_kegg)
print(g_kegg)
ggsave(g_kegg,filename = paste0(pro,'_kegg_up_down.png') )
if(F){
##GSEA
#GSEA算法跟上面的使用差异基因集做超几何分布检验不一样
kk_gse <- gseKEGG(geneList = geneList,
organism = 'hsa',
nPerm = 1000,
minGSSize = 20,
pvalueCutoff = 0.9,
verbose = FALSE)
head(kk_gse)[,1:6]
gseaplot(kk_gse, geneSetID = rownames(kk_gse[1,]))
gseaplot(kk_gse, 'hsa04110',title = 'Cell cycle')
kk=DOSE::setReadable(kk_gse, OrgDb='org.Hs.eg.db',keyType='ENTREZID')
tmp=kk@result
write.csv(kk@result,paste0(pro,'_kegg.gsea.csv'))
#这里找不到显著下调的通路,可以选择调整阈值,或者其它
down_kegg<-kk_gse[kk_gse$pvalue<0.05 & kk_gse$enrichmentScore < 0,];down_kegg$group=-1
up_kegg<-kk_gse[kk_gse$pvalue<0.05 & kk_gse$enrichmentScore > 0,];up_kegg$group=1
g_kegg=kegg_plot(up_kegg,down_kegg)
print(g_kegg)
ggsave(g_kegg,filename = paste0(pro,'_kegg_gsea.png'))
}
}
##GO database analysis
#做GO数据集超几何分布检验分析,重点在结果的可视化及生物学意义的理解
run_go <- function(gene_up,gene_down,pro='test'){
gene_up=unique(gene_up)
gene_down=unique(gene_down)
gene_diff=unique(c(gene_up,gene_down))
g_list=list(gene_up=gene_up,
gene_down=gene_down,
gene_diff=gene_diff)
#因为go数据库非常多基因集,所以运行速度会很慢。
if(T){
go_enrich_results <- lapply( g_list , function(gene) {
lapply( c('BP','MF','CC') , function(ont) {
cat(paste('Now process ',ont ))
ego <- enrichGO(gene = gene,
#universe = gene_all,
OrgDb = org.Hs.eg.db,
ont = ont ,
pAdjustMethod = "BH",
pvalueCutoff = 0.99,
qvalueCutoff = 0.99,
readable = TRUE)
print( head(ego) )
return(ego)
})
})
save(go_enrich_results,file =paste0(pro, '_go_enrich_results.Rdata'))
}
#只有第一次需要运行,就保存结果哈,下次需要探索结果,就载入即可
load(file=paste0(pro, '_go_enrich_results.Rdata'))
n1= c('gene_up','gene_down','gene_diff')
n2= c('BP','MF','CC')
for (i in 1:3){
for (j in 1:3){
fn=paste0(pro, '_dotplot_',n1[i],'_',n2[j],'.png')
cat(paste0(fn,'\n'))
png(fn,res=150,width = 1080)
print( dotplot(go_enrich_results[[i]][[j]] ))
dev.off()
}
}
}
kegg_plot <- function(up_kegg,down_kegg){
dat=rbind(up_kegg,down_kegg)
colnames(dat)
dat$pvalue = -log10(dat$pvalue)
dat$pvalue=dat$pvalue*dat$group
dat=dat[order(dat$pvalue,decreasing = F),]
g_kegg<- ggplot(dat, aes(x=reorder(Description,order(pvalue, decreasing = F)), y=pvalue, fill=group)) +
geom_bar(stat="identity") +
scale_fill_gradient(low="blue",high="red",guide = FALSE) +
scale_x_discrete(name ="Pathway names") +
scale_y_continuous(name ="log10P-value") +
coord_flip() + theme_bw()+theme(plot.title = element_text(hjust = 0.5))+
ggtitle("Pathway Enrichment")
}
下面是对Function的解释:
包装成函数的注释流程(可替换到成Function):
pro='test'
library(ggplot2)
gene_up=unique(gene_up) #unique去重
gene_down=unique(gene_down)
gene_diff=unique(c(gene_up,gene_down))
##over-representation test
#下面把3个基因集分开做超几何分布检验
##上调基因集
kk.up <- enrichKEGG(gene = gene_up,
organism = 'hsa',
#universe = gene_all,
pvalueCutoff = 0.9,
qvalueCutoff =0.9)
head(kk.up)[,1:6]
kk=kk.up
dotplot(kk)
kk=DOSE::setReadable(kk,
OrgDb='org.Hs.eg.db',
keyType='ENTREZID')
write.csv(kk@result,paste0(pro,'_kk.up.csv'))

图 上调基因集
#下调基因集
kk.down <- enrichKEGG(gene = gene_down,
organism = 'hsa',
#universe = gene_all,
pvalueCutoff = 0.9,
qvalueCutoff =0.9)
head(kk.down)[,1:6]
kk=kk.down
dotplot(kk)
kk=DOSE::setReadable(kk,
OrgDb='org.Hs.eg.db',
keyType='ENTREZID')
write.csv(kk@result,paste0(pro,'_kk.down.csv'))
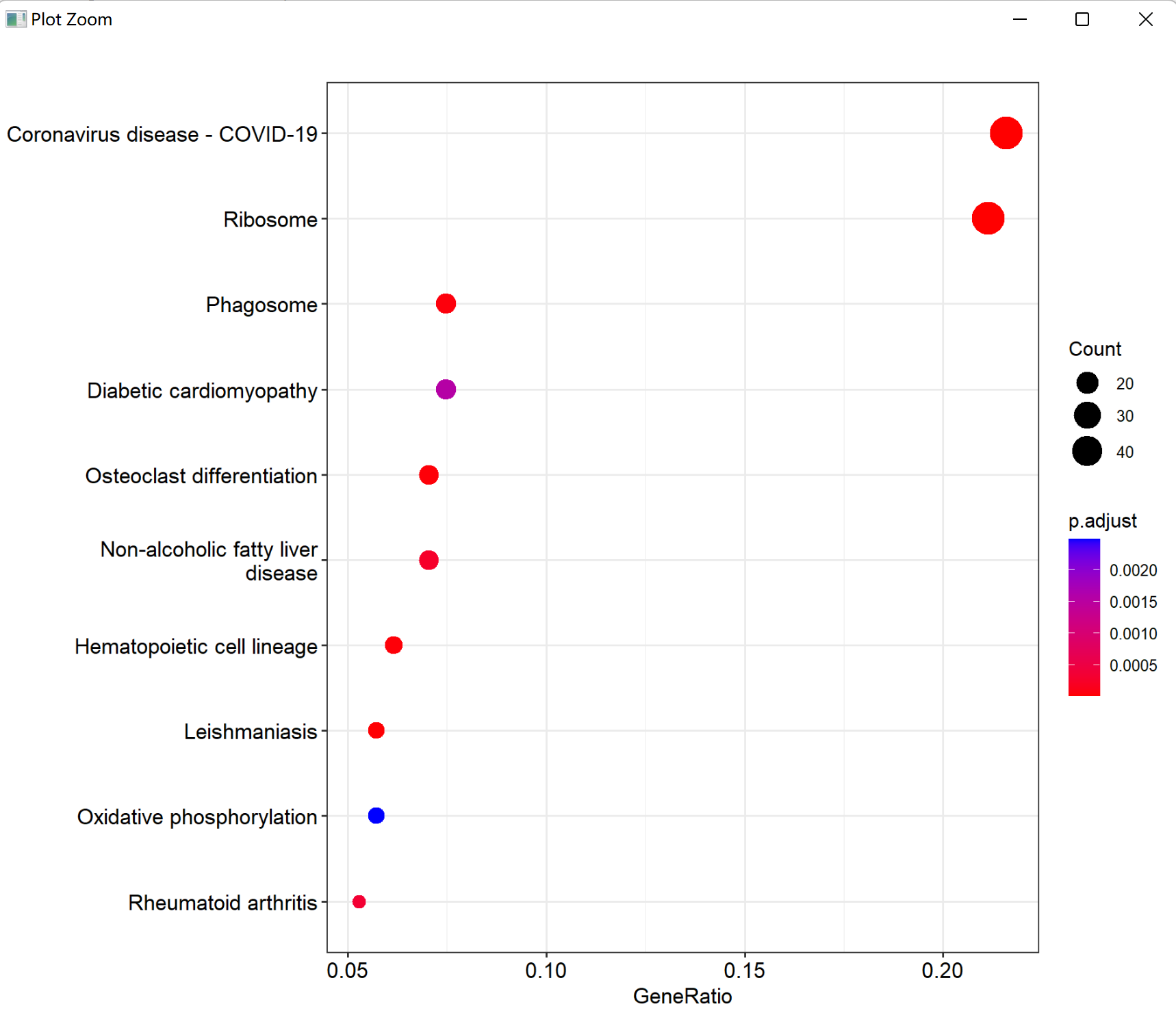
图 下调基因集
#上下调合并后的基因集
kk.diff <- enrichKEGG(gene = gene_diff,
organism = 'hsa',
pvalueCutoff = 0.05)
head(kk.diff)[,1:6]
kk=kk.diff
dotplot(kk)
kk=DOSE::setReadable(kk,
OrgDb='org.Hs.eg.db',
keyType='ENTREZID')
write.csv(kk@result,paste0(pro,'_kk.diff.csv'))

图 上下调合并的基因集
kegg_up_dt <- as.data.frame(kk.up)
kegg_down_dt <- as.data.frame(kk.down)
kegg_diff_dt <- as.data.frame(kk.diff)
up_kegg<-kegg_up_dt[kegg_up_dt$pvalue<0.01,];up_kegg$group=1
down_kegg<-kegg_down_dt[kegg_down_dt$pvalue<0.01,];down_kegg$group=-1
#画图设置, 这个图很丑,大家可以自行修改。
g_kegg=kegg_plot(up_kegg,down_kegg)
print(g_kegg)
ggsave(g_kegg,filename = paste0(pro,'_kegg_up_down.png') )
##GO database analysis
#做GO数据集超几何分布检验分析,重点在结果的可视化及生物学意义的理解
run_go <- function(gene_up,gene_down,pro='test'){
gene_up=unique(gene_up)
gene_down=unique(gene_down)
gene_diff=unique(c(gene_up,gene_down))
g_list=list(gene_up=gene_up,
gene_down=gene_down,
gene_diff=gene_diff)
#因为go数据库非常多基因集,所以运行速度会很慢。
if(T){
go_enrich_results <- lapply( g_list , function(gene) {
lapply( c('BP','MF','CC') , function(ont) {
cat(paste('Now process ',ont ))
ego <- enrichGO(gene = gene,
#universe = gene_all,
OrgDb = org.Hs.eg.db,
ont = ont ,
pAdjustMethod = "BH",
pvalueCutoff = 0.99,
qvalueCutoff = 0.99,
readable = TRUE)
print( head(ego) )
return(ego)
})
})
save(go_enrich_results,file =paste0(pro, '_go_enrich_results.Rdata'))
}
n1= c('gene_up','gene_down','gene_diff')
n2= c('BP','MF','CC')
for (i in 1:3){
for (j in 1:3){
fn=paste0(pro, '_dotplot_',n1[i],'_',n2[j],'.png')
cat(paste0(fn,'\n'))
png(fn,res=150,width = 1080)
print( dotplot(go_enrich_results[[i]][[j]] ))
dev.off()
}
}
kegg_plot <- function(up_kegg,down_kegg){
dat=rbind(up_kegg,down_kegg)
colnames(dat)
dat$pvalue = -log10(dat$pvalue)
dat$pvalue=dat$pvalue*dat$group
dat=dat[order(dat$pvalue,decreasing = F),]
g_kegg<- ggplot(dat, aes(x=reorder(Description,order(pvalue, decreasing = F)), y=pvalue, fill=group)) +
geom_bar(stat="identity") +
scale_fill_gradient(low="blue",high="red",guide = FALSE) +
scale_x_discrete(name ="Pathway names") +
scale_y_continuous(name ="log10P-value") +
coord_flip() + theme_bw()+theme(plot.title = element_text(hjust = 0.5))+
ggtitle("Pathway Enrichment")
}
Function功能解释至此。
#直接利用之前保存的脚本中的代码对正、负相关基因进行kegg富集分析
run_kegg(gene_up,gene_down,pro='test')

#对正相关基因进行富集分析
go <- enrichGO(gene_up,
OrgDb = "org.Hs.eg.db",
ont="all")
library(ggplot2)
library(stringr)
barplot(go,
split="ONTOLOGY")+ facet_grid(ONTOLOGY~., scale="free")+
ggsave('gene_up_GO_all_barplot.png')
go=DOSE::setReadable(go,
OrgDb='org.Hs.eg.db',
keyType='ENTREZID')
write.csv( go@result,file = 'gene_up_GO_all_barplot.csv')
#对负相关基因进行富集分析
go <- enrichGO(gene_down,
OrgDb = "org.Hs.eg.db",
ont="all")
barplot(go,
split="ONTOLOGY")+ facet_grid(ONTOLOGY~., scale="free")+
ggsave('gene_down_GO_all_barplot.png')
go=DOSE::setReadable(go, OrgDb='org.Hs.eg.db',keyType='ENTREZID')
write.csv( go@result,file = 'gene_down_GO_all_barplot.csv')

若有收获,就点个赞吧
0 人点赞
